
Fine Mapping of QTLs/QTNs and Mining of Genes Associated with Race 7 of the Soybean Cercospora sojina by Combining Linkages and GWAS

Abstract
1. Introduction
2. Materials and Methods
2.1. Genetic Material
2.2. Field Experiment
2.3. Inoculation of FLS
2.4. Determination of the Diseased Leaf Area Ratio
2.5. Phenotypic Variation Analysis
2.6. QTL Mapping
2.7. Genome-Wide Association Analysis
2.8. Prediction of Candidate Genes
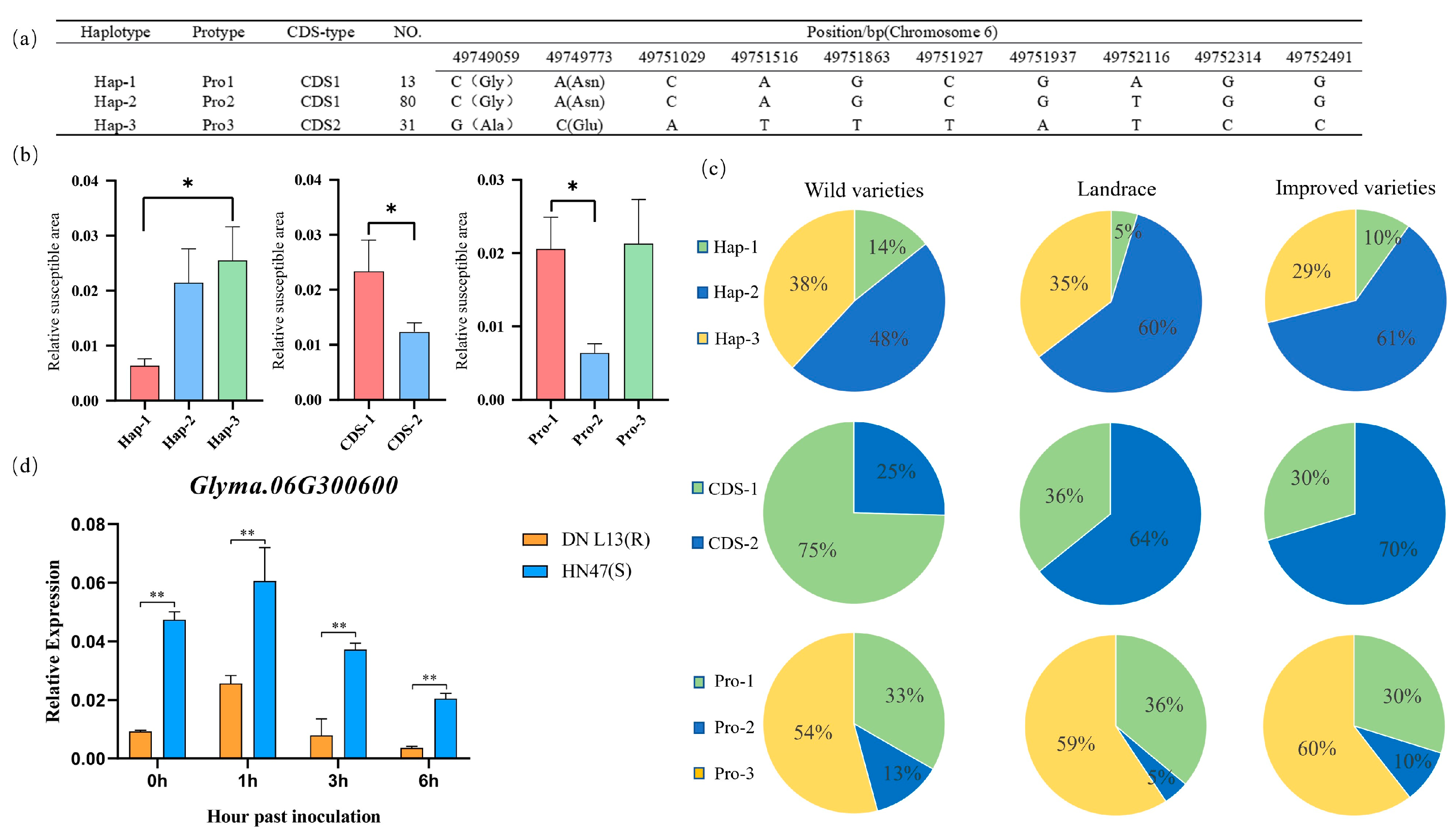
2.9. Analysis of Haplotypes
2.10. Analysis of Candidate Gene Expression
2.11. Analysis of Haplotype Distribution
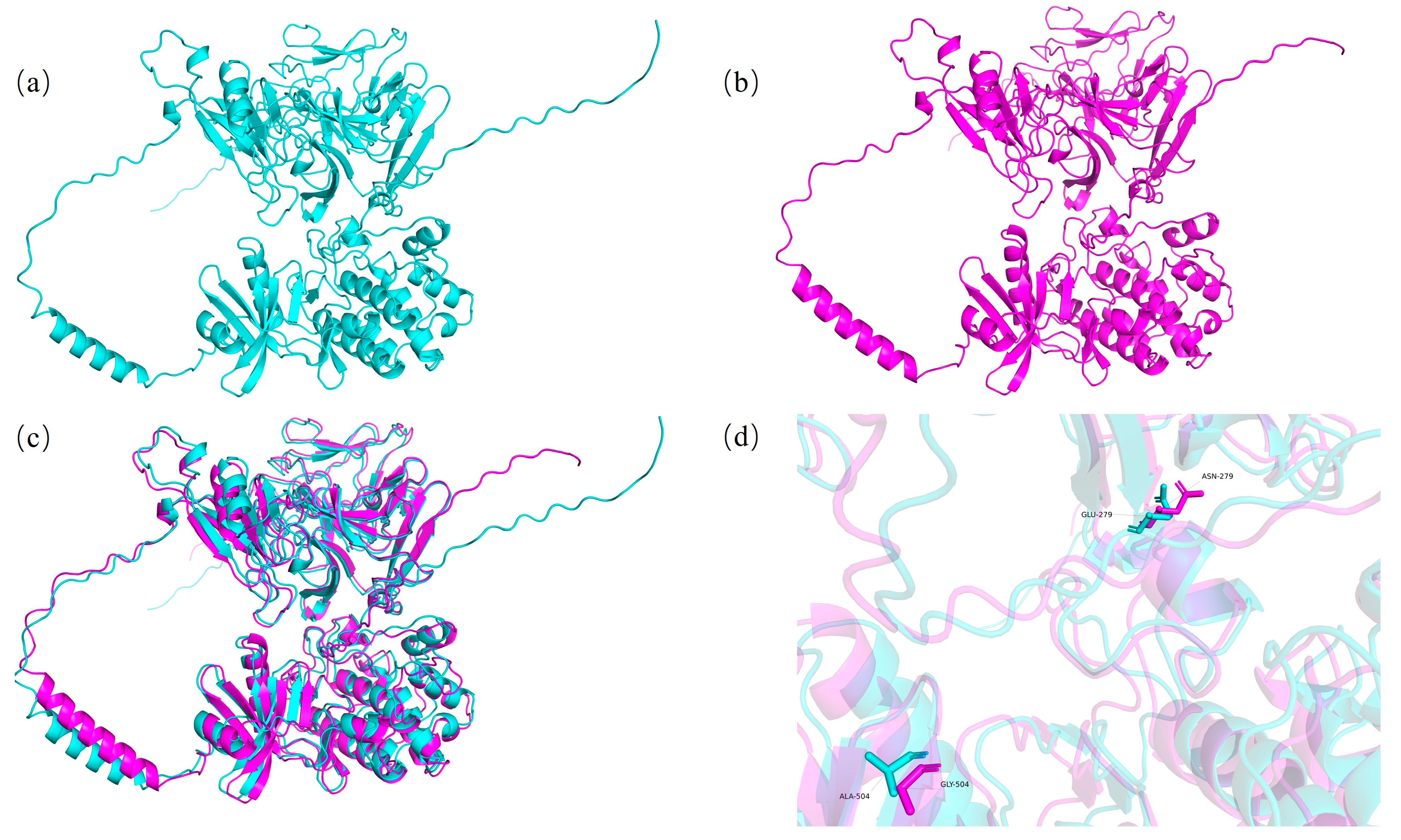
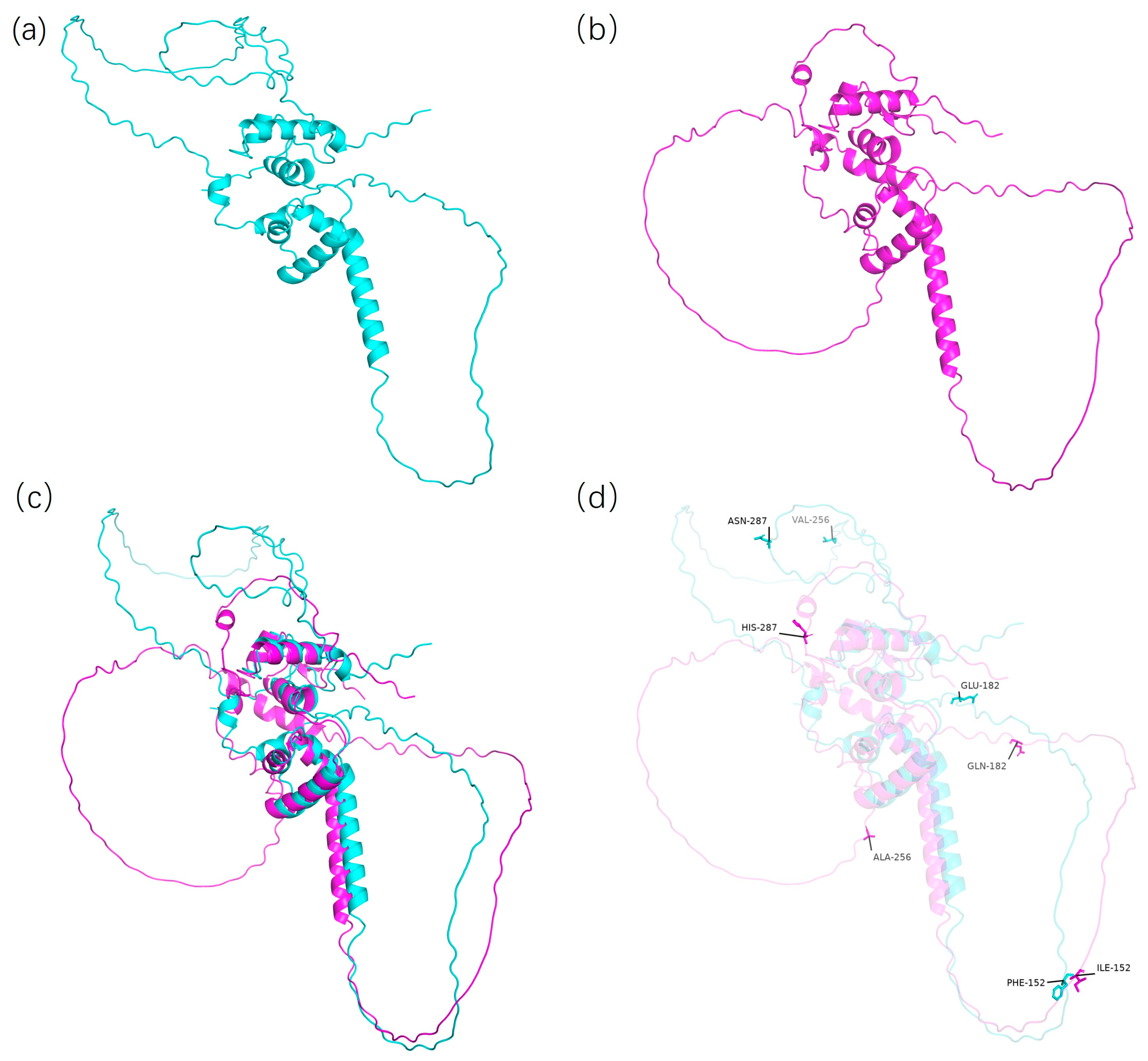
2.12. Methods for Predicting Protein Tertiary Structures Corresponding to Different Haplotypes of Candidate Genes
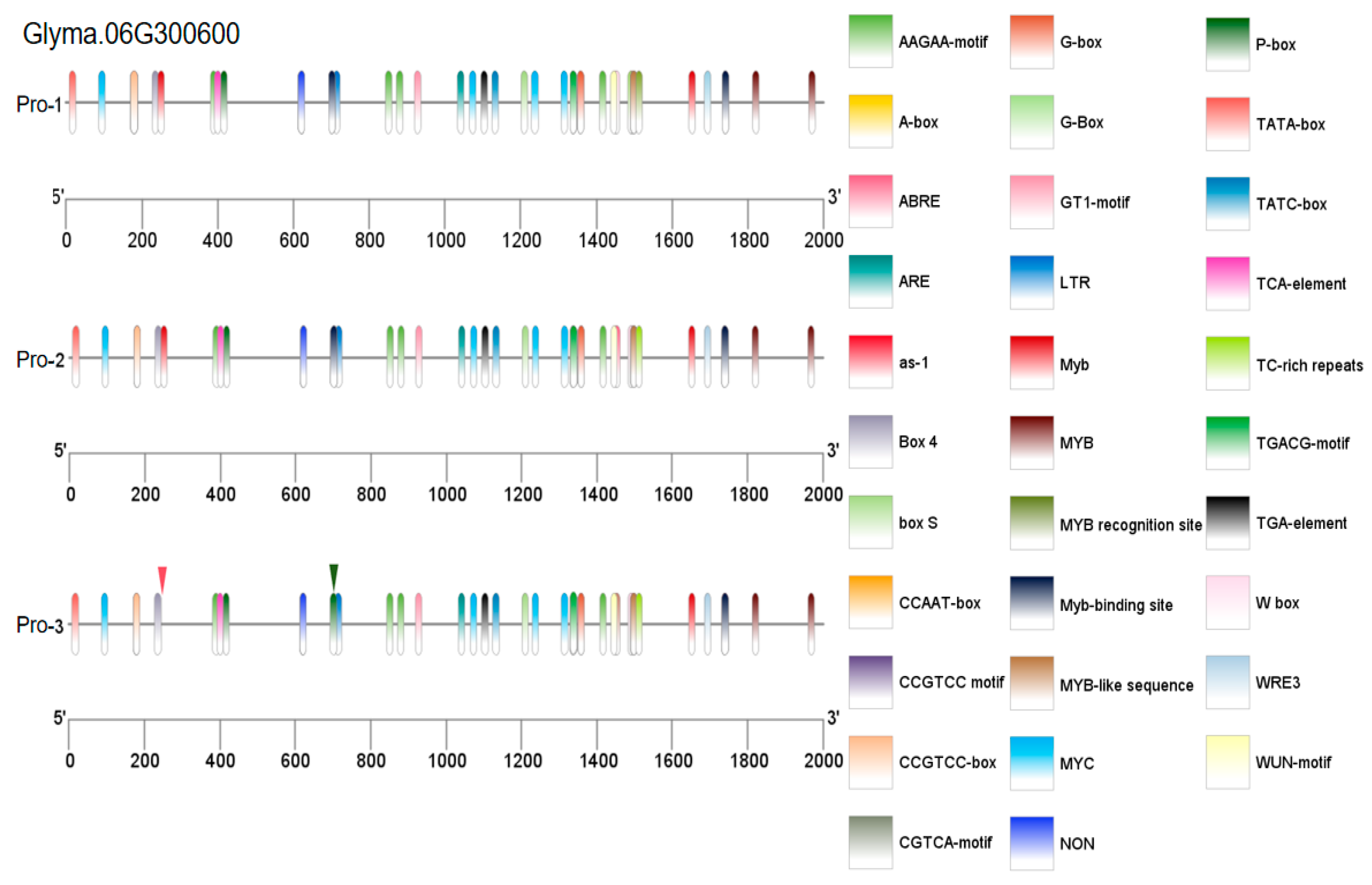
2.13. Promoter Cis-Acting Element Prediction
3. Results and Analysis
3.1. Phenotypic Variation in the PLAD of FLS Race 7 in the Two Populations
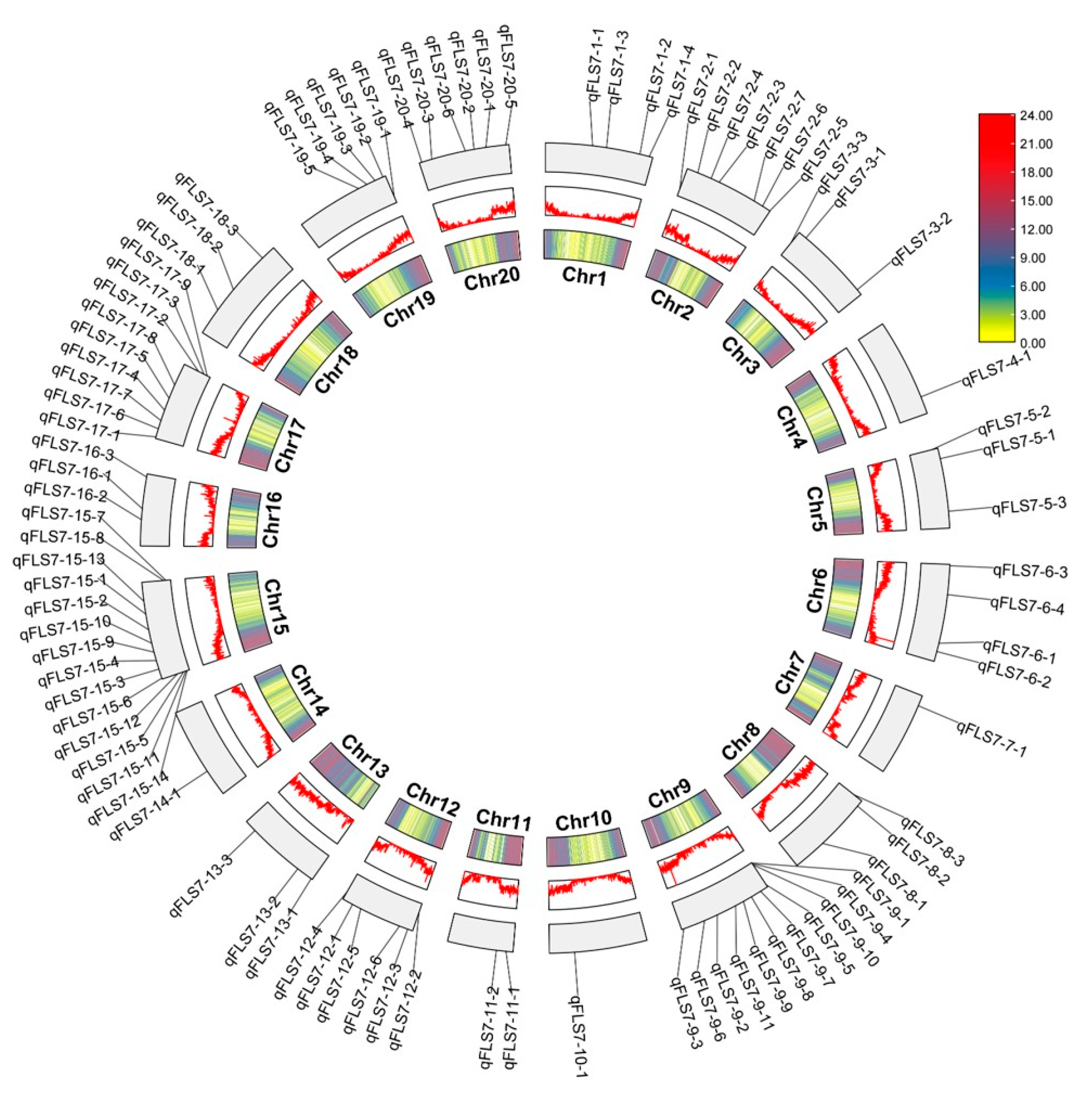
3.2. QTLs for FLS in the RIL3613 Population
3.3. QTN for the PLAD Identified in the GP Population
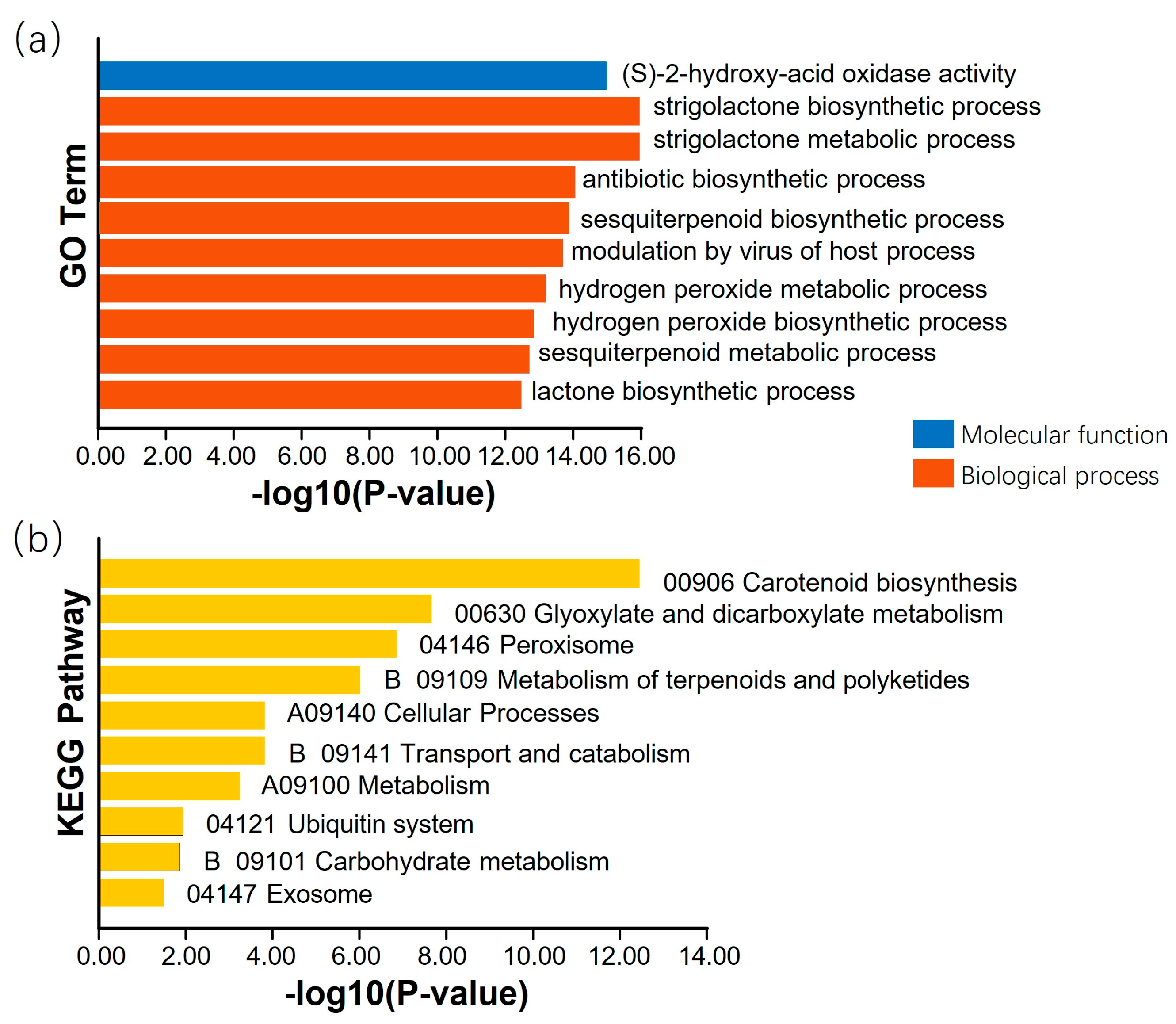
3.4. Candidate Genes for Resistance in FLS Race 7
3.5. Validation of Candidate Genes
3.6. Protein Tertiary Structure Prediction
3.7. Analysis of Promoter Cis-Acting Elements
4. Discussion
4.1. Genetic Characteristics of the PLAD of FLS in Soybean
4.2. Integration of Linkage Analysis and 3VmrMLM Enhances the Accuracy of Mapping Genes Associated with Soybean FLS PLAD
4.3. Colocalization of QTL/QTN with Different Genetic Backgrounds
4.4. Candidate Gene Screening
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Acheng | AC |
| Xiangyang | XY |
| Frogeye leaf spot | FLS |
| Single-seed design method | SSD |
| Quantitative trait loci | QTL |
| Quantitative trait nucleotide | QTN |
| Analysis of variance | ANOVA |
| Percentage of leaf area diseased | PLAD |
References
- Kim, H.; Newell, A.D.; Cota-Sieckmeyer, R.G.; Rupe, J.C.; Fakhoury, A.M.; Bluhm, B.H. Mating-type distribution and genetic diversity of Cercospora sojina populations on soybean from Arkansas: Evidence for potential sexual reproduction. Phytopathology 2013, 103, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Athow, K.; Probst, A.H. The inheritance of resistance to frog-eye leaf spot of soybeans. Phytopathology 1952, 42, 660–662. [Google Scholar]
- Yorinori, J.T. Management of foliar fungal diseases in soybean in Brazil. In Pest Management in Soybean; Copping, L.G., Green, M.B., Rees, R.T., Eds.; Springer: Dordrecht, The Netherlands, 1992; pp. 185–195. [Google Scholar]
- Mian, M.A.R.; Missaoui, A.M.; Walker, D.R.; Phillips, D.V.; Boerma, H.R. Frogeye leaf spot of soybean: A review and proposed race designations for isolates of Cercospora sojina Hara. Crop Sci. 2008, 48, 14–24. [Google Scholar] [CrossRef]
- Soares, A.P.G.; Guillin, E.A.; Borges, L.L.; Silva, A.C.T.d.; Almeida, Á.M.R.d.; Grijalba, P.E.; Gottlieb, A.M.; Bluhm, B.H.; Oliveira, L.O.d. More Cercospora species infect soybeans across the Americas than meets the eye. PLoS ONE 2015, 10, e0133495. [Google Scholar] [CrossRef]
- Phillips, D.V.; Boerma, H.R. Cercospora sojina race 5: A threat to soybeans in the southeastern United States. Phytopathology 1981, 71, 334–336. [Google Scholar] [CrossRef]
- Xiao, G.; Fang, T.; Hou, J.; Wang, W.; Sun, L. Advances in Cercospora sojina physiological races and inheritance of resistance to soybean frogeye leaf spot. J. Plant Genet. Resour. 2024, 25, 870–881. [Google Scholar]
- Probst, A.H.; Athow, K.L.; Laviolette, F.A. Inheritance of resistance to race 2 of Cercospora sojina in soybeans. Crop Sci. 1965, 5, 332. [Google Scholar] [CrossRef]
- Phillips, D.V.; Boerma, H.R. Two Genes for Resistance to Race 5 of Cercospora sojina in Soybeans. Phytopathology 1982, 72, 764–766. [Google Scholar] [CrossRef]
- Yang, W.; Weaver, D.B.; Nielsen, B.L.; Qiu, J.; Salamini, F. Molecular mapping of a new gene for resistance to frogeye leaf spot of soya bean in ‘Peking’. Plant Breed. 2001, 120, 73–78. [Google Scholar] [CrossRef]
- Gu, X.; Huang, S.; Zhu, Z.; Ma, Y.; Yang, X.; Yao, L.; Gao, X.; Zhang, M.; Liu, W.; Qiu, L. Genome-wide association of single nucleotide polymorphism loci and candidate genes for frogeye leaf spot (Cercospora sojina) resistance in soybean. BMC Plant Biol. 2021, 21, 588. [Google Scholar] [CrossRef]
- Liang, X.; Yang, W. Association analysis of resistance of soybean to Cercospora sojina Hara Race 10 based on SSR markers. Chin. J. Oil Crop Sci. 2021, 43, 1132–1140. [Google Scholar]
- Na, C.; Miao, H.; Jiang, H.; Ma, J.; Liu, H.; Lv, S.; Zhou, J.; Yang, Y.; Zhan, Y.; Teng, W. Genome-wide association analysis of resistance to frogeye leaf spot China race 7 in soybean based on high-throughput sequencing. Theor. Appl. Genet. 2023, 136, 119. [Google Scholar] [CrossRef]
- McDonald, S.C.; Buck, J.; Song, Q.; Li, Z. Genome-wide association study reveals novel loci and a candidate gene for resistance to frogeye leaf spot (Cercospora sojina) in soybean. Mol. Genet. Genom. 2023, 298, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, K.; Sun, X.; Huang, S.; Wang, J.; Yang, C.; Siyal, M.; Wang, C.; Guo, C.; Hu, X. Detection of QTL and QTN and candidate genes for oil content in soybean using a combination of four-way-RIL and germplasm populations. Crop J. 2020, 8, 802–811. [Google Scholar] [CrossRef]
- Wang, J.; Hu, B.; Huang, S.; Hu, X.; Siyal, M.; Yang, C.; Zhao, H.; Yang, T.; Li, H.; Hou, Y. SNP-bin linkage analysis and genome-wide association study of plant height in soybean. Crop Pasture Sci. 2022, 73, 222–237. [Google Scholar] [CrossRef]
- Yao, D.; Wang, P.; Zhang, J.; Liu, Z.; Guan, S.; Liu, S.; Qu, J. A QTL mapping analysis of main yield traits in soybean. J. South China Agric. Univ. 2014, 35, 41–46. [Google Scholar]
- Zhang, K.; Liu, S.; Li, W.; Liu, S.; Li, X.; Fang, Y.; Zhang, J.; Wang, Y.; Xu, S.; Zhang, J. Identification of QTNs controlling seed protein content in soybean using multi-locus genome-wide association studies. Front. Plant Sci. 2018, 9, 1690. [Google Scholar] [CrossRef]
- Han, Z.; Ke, H.; Li, X.; Peng, R.; Zhai, D.; Xu, Y.; Wu, L.; Wang, W.; Cui, Y. Detection of epistasis interaction loci for fiber quality-related trait via 3VmrMLM in upland cotton. Front. Plant Sci. 2023, 14, 1250161. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Y.; Xiang, Y.; Liu, M.; Zhang, Y. IIIVmrMLM: The R and C++ tools associated with 3VmrMLM, a comprehensive GWAS method for dissecting quantitative traits. Mol. Plant 2022, 15, 1251–1253. [Google Scholar] [CrossRef]
- Liu, X.; Shang, C.; Duan, P.; Yang, J.; Wang, J.; Sui, D.; Chen, G.; Li, X.; Li, G.; Hu, S. The SlWRKY42–SlMYC2 module synergistically enhances tomato saline–alkali tolerance by activating the jasmonic acid signaling and spermidine biosynthesis pathway. J. Integr. Plant Biol. 2025, 67, 1254–1273. [Google Scholar] [CrossRef]
- Mengiste, T.; Chen, X.; Salmeron, J.; Dietrich, R. The BOTRYTIS SUSCEPTIBLE1 gene encodes an R2R3MYB transcription factor protein that is required for biotic and abiotic stress responses in Arabidopsis. Plant Cell 2003, 15, 2551–2565. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Na, C.; Jing, Y.; Cui, Z.; Li, N.; Zhan, Y.; Teng, W.; Li, Y.; Li, W.; Zhao, X. Genome-wide association analysis and gene mining of resistance to China race 1 of frogeye leaf spot in soybean. Front. Plant Sci. 2022, 13, 867713. [Google Scholar] [CrossRef]
- Pham, A.T.; Harris, D.K.; Buck, J.; Hoskins, A.; Serrano, J.; Abdel-Haleem, H.; Cregan, P.; Song, Q.; Boerma, H.R.; Li, Z. Fine mapping and characterization of candidate genes that control resistance to Cercospora sojina K. Hara in two soybean germplasm accessions. PLoS ONE 2015, 10, e0126753. [Google Scholar] [CrossRef]
- Gao, Y.; Zhu, J. Advance on methodology of QTL mapping for plants. Hereditas 2000, 22, 175–179. [Google Scholar]
- Yan, H.; Wang, H.; Cheng, H.; Hu, Z.; Chu, S.; Zhang, G.; Yu, D. Detection and fine-mapping of SC7 resistance genes via linkage and association analysis in soybean. J. Integr. Plant Biol. 2015, 57, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, P.; Zhang, K.; Liu, S.; Qi, Z.; Fang, Y.; Wang, Y.; Tian, X.; Song, J.; Wang, J. Fine mapping QTL and mining genes for protein content in soybean by the combination of linkage and association analysis. Theor. Appl. Genet. 2021, 134, 1095–1122. [Google Scholar] [CrossRef]
- Sun, M.; Zhao, T.; Liu, S.; Han, J.; Wang, Y.; Zhao, X.; Li, Y.; Teng, W.; Zhan, Y.; Han, Y. QTL Detection of Salt Tolerance at Soybean Seedling Stage Based on Genome-Wide Association Analysis and Linkage Analysis. Plants 2024, 13, 2283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, S.; Wu, X.; Han, L.; Wang, Y.; Wen, Y. Identification of QTNs, QTN-by-environment interactions and genes for yield-related traits in rice using 3VmrMLM. Front. Plant Sci. 2022, 13, 995609. [Google Scholar] [CrossRef]
- Xiong, X.; Li, J.; Su, P.; Duan, H.; Sun, L.; Xu, S.; Sun, Y.; Zhao, H.; Chen, X.; Ding, D. Genetic dissection of maize (Zea mays L.) chlorophyll content using multi-locus genome-wide association studies. BMC Genom. 2023, 24, 384. [Google Scholar] [CrossRef]
- Niu, H.; Kuang, M.; Huang, L.; Shang, H.; Yuan, Y.; Ge, Q. Lint percentage and boll weight QTLs in three excellent upland cotton (Gossypium hirsutum): ZR014121, CCRI60, and EZ60. BMC Plant Biol. 2023, 23, 179. [Google Scholar] [CrossRef]
- Hong, H.; Li, M.; Chen, Y.; Wang, H.; Wang, J.; Guo, B.; Gao, H.; Ren, H.; Yuan, M.; Han, Y. Genome-wide association studies for soybean epicotyl length in two environments using 3VmrMLM. Front. Plant Sci. 2022, 13, 1033120. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wang, H.; Sui, Y.; Miao, Y.; Jin, C.; Luo, J. Genome-wide association studies of five free amino acid levels in rice. Front. Plant Sci. 2022, 13, 1048860. [Google Scholar] [CrossRef]
- Wei, N.; Zhang, S.; Liu, Y.; Wang, J.; Wu, B.; Zhao, J.; Qiao, L.; Zheng, X.; Wang, J.; Zheng, J. Genome-wide association study of coleoptile length with Shanxi wheat. Front. Plant Sci. 2022, 13, 1016551. [Google Scholar] [CrossRef] [PubMed]
- Sonah, H.; O’Donoughue, L.; Cober, E.; Rajcan, I.; Belzile, F. Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol. J. 2015, 13, 211–221. [Google Scholar] [CrossRef]
- Hongjiao, J.; Huiyan, Z.; Chunyu, L.; Xiaoyu, L.; Shumei, M.; Yang, W. Soybean variety resource resistant to Cercospora sojina race 7: Mining of elite alleles. Chin. Agric. Sci. Bull. 2020, 36, 97–102. [Google Scholar]
- McAllister, K.R.; Lee, Y.C.; Kantartzi, S.K. QTL mapping for resistance to Cercospora sojina in Essex Forrest soybean (Glycine max L.) lines. J. Plant Breed. Crop Sci. 2021, 13, 14–22. [Google Scholar]
- Dong, Y.; Lai, W.; Ding, J.; Wang, W.; Yu, Z.; Li, Z.; Yang, X.; Liu, L. Excavation the resistance locus to Cercospora sojina Hara race 12 in soybean by association analysis. Soybean Sci. 2016, 35, 402–406. [Google Scholar]
- Stracke, R.; Werber, M.; Weisshaar, B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 2001, 4, 447–456. [Google Scholar] [CrossRef]
- Rabinowicz, P.D.; Braun, E.L.; Wolfe, A.D.; Bowen, B.; Grotewold, E. Maize R2R3 Myb genes: Sequence analysis reveals amplification in the higher plants. Genetics 1999, 153, 427–444. [Google Scholar] [CrossRef]
- Liu, L.; Du, H.; Tang, X.; Wu, Y.; Huang, Y.; Tang, Y. The roles of MYB transcription factors on plant defense responses and its molecular mechanism. Hereditas 2008, 30, 1265–1271. [Google Scholar] [CrossRef]
- Pal, D.; De, K.; Shanks, C.M.; Feng, K.; Yates, T.B.; Morrell-Falvey, J.; Davidson, R.B.; Parks, J.M.; Muchero, W. Core cysteine residues in the Plasminogen-Apple-Nematode (PAN) domain are critical for HGF/c-MET signaling. Commun. Biol. 2022, 5, 646. [Google Scholar] [CrossRef] [PubMed]
- De, K.; Pal, D.; Shanks, C.M.; Yates, T.B.; Feng, K.; Jawdy, S.S.; Hassan, M.M.; Prabhakar, P.K.; Yang, J.Y.; Chapla, D. The Plasminogen-Apple-Nematode (PAN) domain suppresses JA/ET defense pathways in plants. bioRxiv 2023. [Google Scholar] [CrossRef]
- Freitas, C.D.T.; Costa, J.H.; Germano, T.A.; Rocha, R.d.O.; Ramos, M.V.; Bezerra, L.P. Class III plant peroxidases: From classification to physiological functions. Int. J. Biol. Macromol. 2024, 263, 130306. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environment | DN L13 (Average) | Hh 36 (Average) | Average | Standard Deviation | Minimum | Maximum | Skewness | Kurtosis |
|---|---|---|---|---|---|---|---|---|
| XY | 0.015 | 0.009 | 0.007 | 0.014 | 0.000 | 0.114 | 5.238 | 35.731 |
| AC | 0.038 | 0.000 | 0.010 | 0.016 | 0.000 | 0.106 | 3.371 | 15.099 |
| Environment | Average | Standard Deviation | Minimum | Maximum | Skewness | Kurtosis |
|---|---|---|---|---|---|---|
| XY | 0.031 | 0.027 | 0 | 0.163 | 1.224 | 1.398 |
| AC | 0.027 | 0.022 | 0 | 0.103 | 1.092 | 0.404 |
| QTL | Eve. | Chromosome | Position | LeftMarker | RightMarker | LOD | PVE (%) | Add |
|---|---|---|---|---|---|---|---|---|
| qFLS7-2–2 | XY | 2 | 30 | 36c02048 | 36c02047 | 10.3684 | 0.3497 | −0.0454 |
| AC | 2 | 30 | 36c02048 | 36c02047 | 7.166 | 1.3612 | −0.0301 | |
| Ave | 2 | 30 | 36c02048 | 36c02047 | 6.2352 | 0.585 | −0.0233 | |
| qFLS7-2–4 | XY | 2 | 92 | 36c02078 | 36c02085 | 9.1225 | 0.3846 | −0.0344 |
| AC | 2 | 92 | 36c02078 | 36c02085 | 4.3503 | 2.0034 | −0.016 | |
| Ave | 2 | 92 | 36c02078 | 36c02085 | 4.7828 | 0.6514 | −0.0157 | |
| qFLS7-9–4 | XY | 9 | 75 | 36c09006 | 36c09011 | 11.9232 | 0.387 | −0.0342 |
| AC | 9 | 75 | 36c09006 | 36c09011 | 9.0694 | 2.1026 | −0.0152 | |
| Ave | 9 | 75 | 36c09006 | 36c09011 | 9.1487 | 0.6636 | −0.0145 | |
| qFLS7-9–6 | XY | 9 | 129 | 36c09139 | 36c09078 | 12.3221 | 0.3774 | −0.0349 |
| AC | 9 | 129 | 36c09139 | 36c09078 | 8.3433 | 2.0573 | −0.0155 | |
| Ave | 9 | 129 | 36c09139 | 36c09078 | 7.8439 | 0.6294 | −0.0179 | |
| qFLS7-13–2 | XY | 13 | 95 | 36c13012 | 36c13013 | 8.1551 | 0.3872 | −0.0342 |
| AC | 13 | 96 | 36c13012 | 36c13013 | 5.3459 | 1.9547 | −0.0122 | |
| Ave | 13 | 96 | 36c13012 | 36c13013 | 6.9489 | 0.7106 | −0.0167 | |
| qFLS7-14–1 | XY | 14 | 41 | 36c14093 | 36c14072 | 16.1204 | 0.3498 | −0.0455 |
| Ave | 14 | 41 | 36c14093 | 36c14072 | 11.992 | 0.5857 | −0.0233 | |
| AC | 14 | 42 | 36c14093 | 36c14072 | 9.0625 | 1.9965 | −0.0164 | |
| qFLS7-15–5 | XY | 15 | 388 | 36c15009 | 36c15010 | 9.0114 | 0.3662 | −0.0382 |
| AC | 15 | 388 | 36c15009 | 36c15010 | 11.0052 | 2.1013 | −0.0156 | |
| Ave | 15 | 388 | 36c15009 | 36c15010 | 6.2068 | 0.6145 | −0.0151 | |
| qFLS7-17–3 | XY | 17 | 89 | 36c17083 | 36c17082 | 13.9472 | 0.367 | −0.038 |
| AC | 17 | 89 | 36c17083 | 36c17082 | 10.3001 | 2.2343 | −0.0148 | |
| Ave | 17 | 90 | 36c17083 | 36c17082 | 9.6205 | 0.5853 | −0.0233 | |
| qFLS7-17–4 | XY | 17 | 95 | 36c17016 | 36c17017 | 11.4424 | 0.3598 | −0.0369 |
| Ave | 17 | 95 | 36c17016 | 36c17017 | 7.0466 | 0.5835 | −0.0204 | |
| AC | 17 | 96 | 36c17016 | 36c17017 | 7.4173 | 1.3612 | −0.0301 |
| Environ- ment | Marker | Chromo- some | Position (bp) | LOD (Q) | Add | LOD (QE) | Add * env1 | Add * env2 | r2 (%) | p Value | Signi- ficance |
|---|---|---|---|---|---|---|---|---|---|---|---|
| XY | AX-90327525 | 3 | 25,283 | 6.67 | −0.0017 | 5.08 | 2.14 × 10−7 | SIG | |||
| XY | AX-90470366 | 3 | 44,602,404 | 7.25 | −0.0022 | 5.32 | 5.58 × 10−8 | SIG | |||
| XY | AX-90383019 | 4 | 12,560,483 | 5.49 | −0.0004 | 4.53 | 3.23 × 10−6 | SUG | |||
| XY | AX-90524088 | 6 | 48,913,997 | 10.14 | −0.002 | 7.82 | 7.27 × 10−11 | SIG | |||
| XY | AX-90443776 | 9 | 39,781,488 | 4.48 | −0.0036 | 2.21 | 3.30 × 10−5 | SUG | |||
| XY | AX-90430118 | 13 | 28,665,638 | 9.28 | 0.0026 | 6.94 | 5.23 × 10−10 | SIG | |||
| XY | AX-90524893 | 13 | 30,790,917 | 5.65 | −0.0003 | 4.06 | 2.24 × 10−6 | SUG | |||
| XY | AX-90439650 | 13 | 34,989,937 | 5.63 | −0.0011 | 4.26 | 2.36 × 10−6 | SUG | |||
| XY | AX-90508257 | 16 | 31,840,753 | 6.26 | −0.0046 | 4.78 | 5.55 × 10−7 | SIG | |||
| AC | AX-90468600 | 4 | 37,488,739 | 19.83 | −0.001 | 6.80 | 1.47 × 10−20 | SIG | |||
| AC | AX-90332112 | 8 | 7,608,204 | 16.58 | −0.0076 | 1.42 | 2.62 × 10−17 | SIG | |||
| AC | AX-90506929 | 9 | 34,223,245 | 4.70 | 0.0024 | 1.04 | 1.98 × 10−5 | SUG | |||
| AC | AX-90463766 | 14 | 43,424,209 | 8.69 | 0.0006 | 2.56 | 2.05 × 10−9 | SIG | |||
| AC | AX-90517572 | 17 | 25,347,969 | 40.20 | 0.0002 | 16.5 | 6.40 × 10−41 | SIG | |||
| AC | AX-90437152 | 17 | 36,392,027 | 82.71 | 0.0002 | 49.1 | 1.96 × 10−83 | SIG | |||
| AC | AX-90433524 | 18 | 6,078,523 | 3.01 | −0.0001 | 0.94 | 0.001 | SUG | |||
| Joint | AX-90325055 | 2 | 14,798,048 | 4.88 | 0.0032 | 1.35 | 1.31 × 10−5 | SUG | |||
| Joint | AX-90468600 | 4 | 37,488,739 | 11.13 | −0.0001 | 3.07 | 7.44 × 10−12 | SIG | |||
| Joint | AX-90319684 | 5 | 32,124,284 | 8.54 | 0.0011 | 2.26 | 2.90 × 10−9 | SIG | |||
| Joint | AX-90517572 | 17 | 25,347,969 | 16.94 | 0.0015 | 4.97 | 1.15 × 10−17 | SIG | |||
| Joint | AX-90437152 | 17 | 36,392,027 | 51.02 | −0.0003 | 17.6 | 9.49 × 10−52 | SIG | |||
| Joint | AX-90468600 | 4 | 37,488,739 | 5.5474 | −0.0007 | 0.0007 | 1.51 | 2.84 × 10−6 | SUG | ||
| Joint | AX-90339041 | 8 | 7,384,418 | 4.2897 | −0.0001 | 0.0001 | 1.12 | 5.13 × 10−5 | SUG | ||
| Joint | AX-90506929 | 9 | 34,223,245 | 9.0559 | 0.0027 | −0.0027 | 2.37 | 8.80 × 10−10 | SIG | ||
| Joint | AX-90444106 | 13 | 1,163,621 | 4.3602 | 0.0031 | −0.0031 | 1.14 | 4.36 × 10−5 | SUG | ||
| Joint | AX-90412520 | 14 | 5,629,805 | 7.4253 | −0.0007 | 0.0007 | 1.96 | 3.76 × 10−8 | SIG | ||
| Joint | AX-90517572 | 17 | 25,347,969 | 21.0304 | −0.0004 | 0.0004 | 6.27 | 9.34 × 10−22 | SIG | ||
| Joint | AX-90437152 | 17 | 36,392,027 | 52.8551 | 0.0006 | −0.0006 | 18.4 | 1.40 × 10−53 | SIG |
| QTN | Enviro- nment | Chromo- some | Position (bp) | r2 (%) | p Value | QTL | LeftMarker | RightMarker |
|---|---|---|---|---|---|---|---|---|
| AX-90325055 | Joint | 2 | 14,798,048 | 1.346 | 1.30654 × 10−5 | qFLS7-2–4 | 14,075,423 | 14,807,628 |
| AX-90319684 | Joint | 5 | 32,124,284 | 2.2618 | 2.89977 × 10−9 | qFLS7-5–1 | 5,605,773 | 34,482,017 |
| AX-90524088 | XY | 6 | 48,913,997 | 7.824 | 7.27444 × 10−11 | qFLS7-6–1 | 45,980,403 | 50,219,401 |
| AX-90332112 | AC | 8 | 7,608,204 | 1.4221 | 2.61598 × 10−17 | qFLS7-8–3 | 2,730,666 | 44,849,884 |
| AX-90339041 | Joint | 8 | 7,384,418 | 1.1162 | 5.1345 × 10−5 | qFLS7-8–3 | 2,730,666 | 44,849,884 |
| AX-90443776 | XY | 9 | 39,781,488 | 2.2125 | 3.29892 × 10−5 | qFLS7-9–6 | 27,334,487 | 48,719,846 |
| AX-90506929 | AC | 9 | 34,223,245 | 1.0424 | 1.97962 × 10−5 | qFLS7-9–6 | 27,334,487 | 48,719,846 |
| AX-90506929 | Joint | 9 | 34,223,245 | 2.3736 | 8.799 × 10−10 | qFLS7-9–6 | 27,334,487 | 48,719,846 |
| AX-90443776 | XY | 9 | 39,781,488 | 2.2125 | 3.29892 × 10−5 | qFLS7-9–5 | 6,963,595 | 38,517,945 |
| AX-90506929 | AC | 9 | 34,223,245 | 1.0424 | 1.97962 × 10−5 | qFLS7-9–5 | 6,963,595 | 38,517,945 |
| AX-90443776 | XY | 9 | 39,781,488 | 2.2125 | 3.29892 × 10−5 | qFLS7-9–2 | 22,385,933 | 43,970,874 |
| AX-90506929 | AC | 9 | 34,223,245 | 1.0424 | 1.97962 × 10−5 | qFLS7-9–2 | 22,385,933 | 43,970,874 |
| AX-90430118 | XY | 13 | 28,665,638 | 6.937 | 5.23201 × 10−10 | qFLS7-13–1 | 1,712,042 | 41,981,042 |
| AX-90524893 | XY | 13 | 30,790,917 | 4.056 | 2.23592 × 10−6 | qFLS7-13–1 | 1,712,042 | 41,981,042 |
| AX-90439650 | XY | 13 | 34,989,937 | 4.2636 | 2.36049 × 10−6 | qFLS7-13–1 | 1,712,042 | 41,981,042 |
| AX-90463766 | AC | 14 | 43,424,209 | 2.562 | 2.05101 × 10−9 | qFLS7-14–1 | 14,348,437 | 45,451,538 |
| AX-90517572 | Joint | 17 | 25,347,969 | 4.9732 | 1.14724 × 10−17 | qFLS7-17–8 | 15,210,682 | 39,468,676 |
| AX-90437152 | Joint | 17 | 36,392,027 | 17.62 | 9.49252 × 10−52 | qFLS7-17–8 | 15,210,682 | 39,468,676 |
| AX-90433524 | AC | 18 | 6,078,523 | 0.9448 | 0.00097409 | qFLS7-18–1 | 4,883,393 | 54,664,500 |
| Genes | Location | Annotation |
|---|---|---|
| Glyma.05G128200 | Chr5:32158033..32162343 | LRR RECEPTOR-LIKE SERINE/THREONINE-PROTEIN KINASE FLS2 |
| Glyma.06G300000 | Chr6:48880588..48882272 | MYB-LIKE DNA-BINDING PROTEIN MYB |
| Glyma.06G300100 | Chr6:48890857..48892413 | MYB-LIKE DNA-BINDING PROTEIN MYB |
| Glyma.06G300200 | Chr6:48908957..48910448 | MYB-LIKE DNA-BINDING PROTEIN MYB |
| Glyma.06G300300 | Chr6:48918302..48920771 | MYB-LIKE DNA-BINDING PROTEIN MYB |
| Glyma.06G300400 | Chr6:48929743..48931574 | MYB-LIKE DNA-BINDING PROTEIN MYB |
| Glyma.06G300600 | Chr6:48955700..48958858 | Protein kinase domain (Pkinase)//S-locus glycoprotein domain (S_locus_glycop)//D-mannose binding lectin (B_lectin)//PAN-like domain (PAN_2) |
| Glyma.08G099400 | Chr8:7597974..7602099 | CBL-INTERACTING SERINE/THREONINE-PROTEIN KINASE 23 |
| Glyma.13G172300 | Chr13:28629837..28635052 | PEROXIDASE 19 |
| Glyma.13G172500 | Chr13:28644818..28649188 | ZINC FINGER FYVE DOMAIN CONTAINING PROTEIN |
| Glyma.13G172900 | Chr13:28697099..28703351 | TRANSCRIPTION FACTOR VOZ1 |
| Glyma.13G194500 | Chr13:30763822..30769223 | LEUCINE-RICH REPEAT-CONTAINING PROTEIN |
| Glyma.13G239100 | Chr13:34957147..34964627 | UBIQUITIN-CONJUGATING ENZYME E2 24-RELATED |
| Glyma.13G239300 | Chr13:34970871..34976701 | CALLOSE SYNTHASE 11 |
| Glyma.17G216100 | Chr17:36392667..36394305 | ETHYLENE-RESPONSIVE TRANSCRIPTION FACTOR ERF003 |
| Gene | Position/bp (Chromosome 6) | Quantity | |||
|---|---|---|---|---|---|
| 49,672,979 | 49,673,106 | 49,674,296 | 49,674,305 | ||
| Glyma.06G300000 | G(Arg) | G(His) | A(Val) | G(Arg) | 33 |
| A(Leu) | G(His) | A(Val) | G(Arg) | 90 | |
| A(Leu) | A(Tyr) | G(Ala) | C(Ser) | 18 | |
| 49,683,236 | 49,683,328 | 49,683,551 | 49,683,641 | ||
| Glyma.06G300100 | G(His) | G(Ala) | G(Gln) | T(Lle) | 68 |
| T(Asn) | A(Val) | C(Glu) | A(Phe) | 56 | |
| 49,749,059 | 49,749,773 | ||||
| Glyma.06G300600 | C(Gly) | A(Asn) | 100 | ||
| G(Ala) | C(Glu) | 32 | |||
| 7,696,683 | |||||
| Glyma.08G099400 | G(Gly) | 152 | |||
| T(Cys) | 2 | ||||
| 29,825,592 | |||||
| Glyma.13G172500 | C(Val) | 147 | |||
| T(Lle) | 1 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Hu, B.; Yu, A.; Liu, Y.; Xu, P.; Wang, Y.; Ding, J.; Zhang, S.; Li, W.-X.; Ning, H. Fine Mapping of QTLs/QTNs and Mining of Genes Associated with Race 7 of the Soybean Cercospora sojina by Combining Linkages and GWAS. Plants 2025, 14, 1988. https://doi.org/10.3390/plants14131988
Liu Y, Hu B, Yu A, Liu Y, Xu P, Wang Y, Ding J, Zhang S, Li W-X, Ning H. Fine Mapping of QTLs/QTNs and Mining of Genes Associated with Race 7 of the Soybean Cercospora sojina by Combining Linkages and GWAS. Plants. 2025; 14(13):1988. https://doi.org/10.3390/plants14131988
Chicago/Turabian StyleLiu, Yanzuo, Bo Hu, Aitong Yu, Yuxi Liu, Pengfei Xu, Yang Wang, Junjie Ding, Shuzhen Zhang, Wen-Xia Li, and Hailong Ning. 2025. "Fine Mapping of QTLs/QTNs and Mining of Genes Associated with Race 7 of the Soybean Cercospora sojina by Combining Linkages and GWAS" Plants 14, no. 13: 1988. https://doi.org/10.3390/plants14131988
APA StyleLiu, Y., Hu, B., Yu, A., Liu, Y., Xu, P., Wang, Y., Ding, J., Zhang, S., Li, W.-X., & Ning, H. (2025). Fine Mapping of QTLs/QTNs and Mining of Genes Associated with Race 7 of the Soybean Cercospora sojina by Combining Linkages and GWAS. Plants, 14(13), 1988. https://doi.org/10.3390/plants14131988