
The Structural Deciphering of the α3 Helix Within ZmHsfA2’S DNA-Binding Domain for the Recognition of Heat Shock Elements in Maize
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
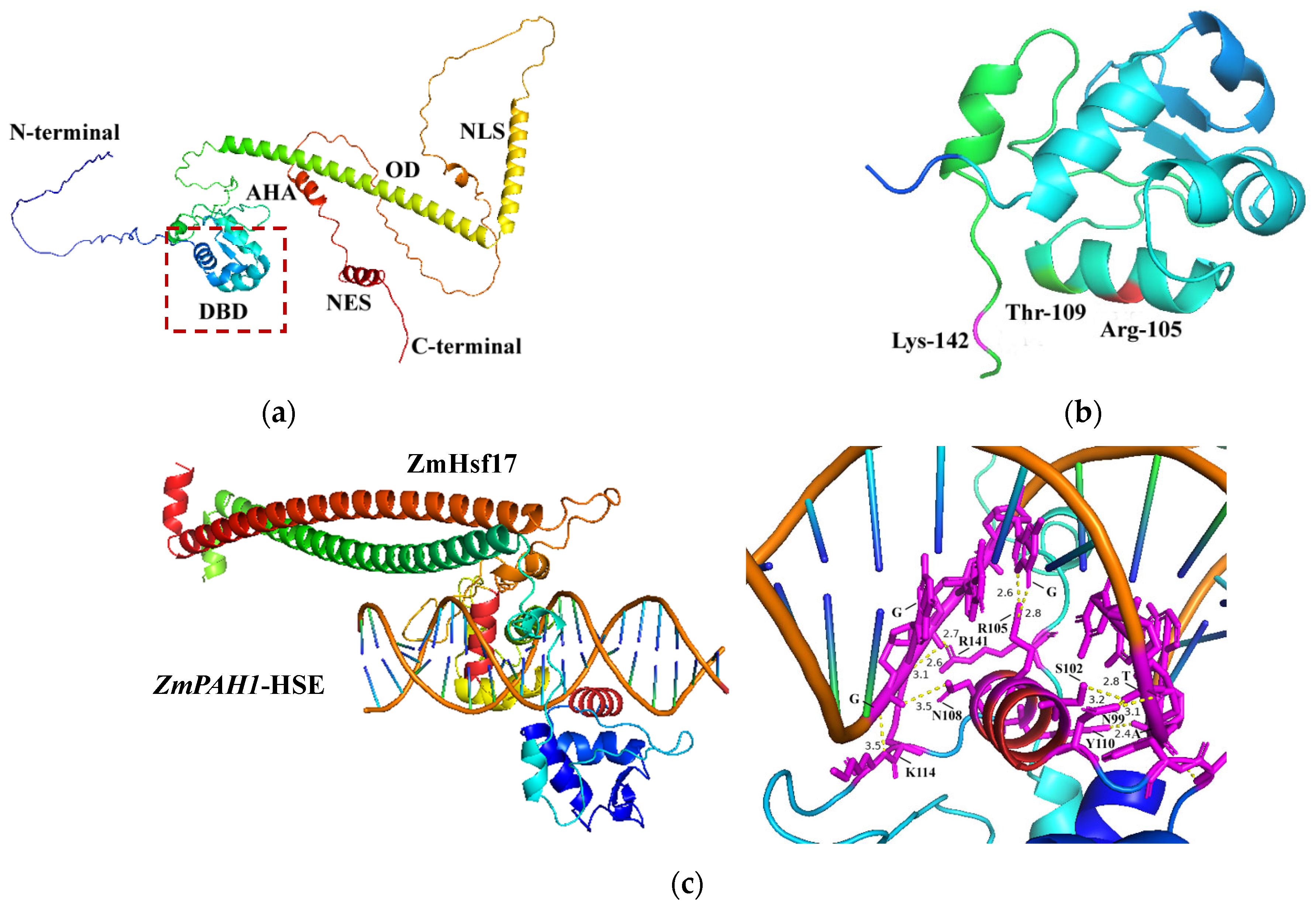
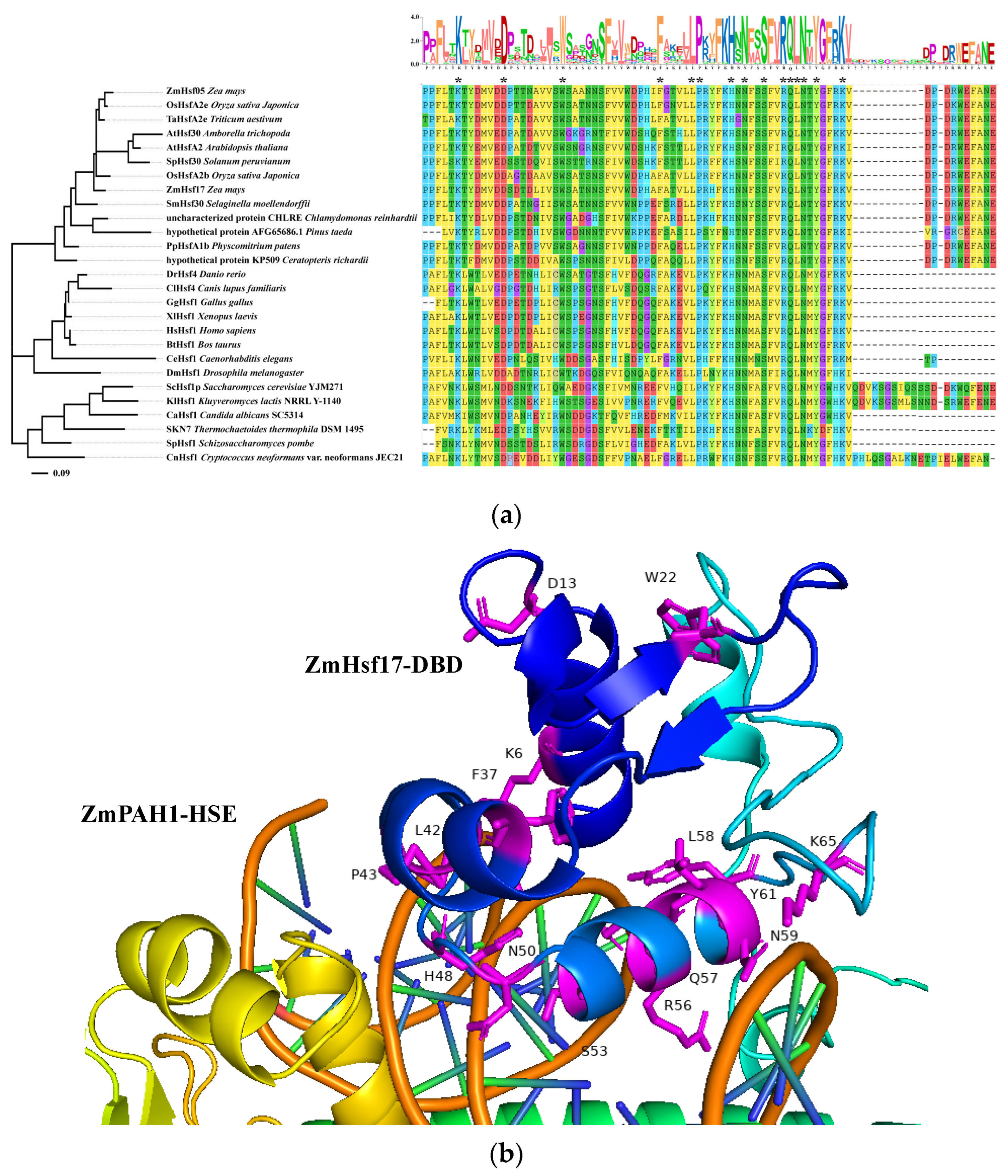
2.1. Modeling Interaction Interfaces of ZmHsf17-DBD and ZmPAH1-HSE Using AlphaFold 3
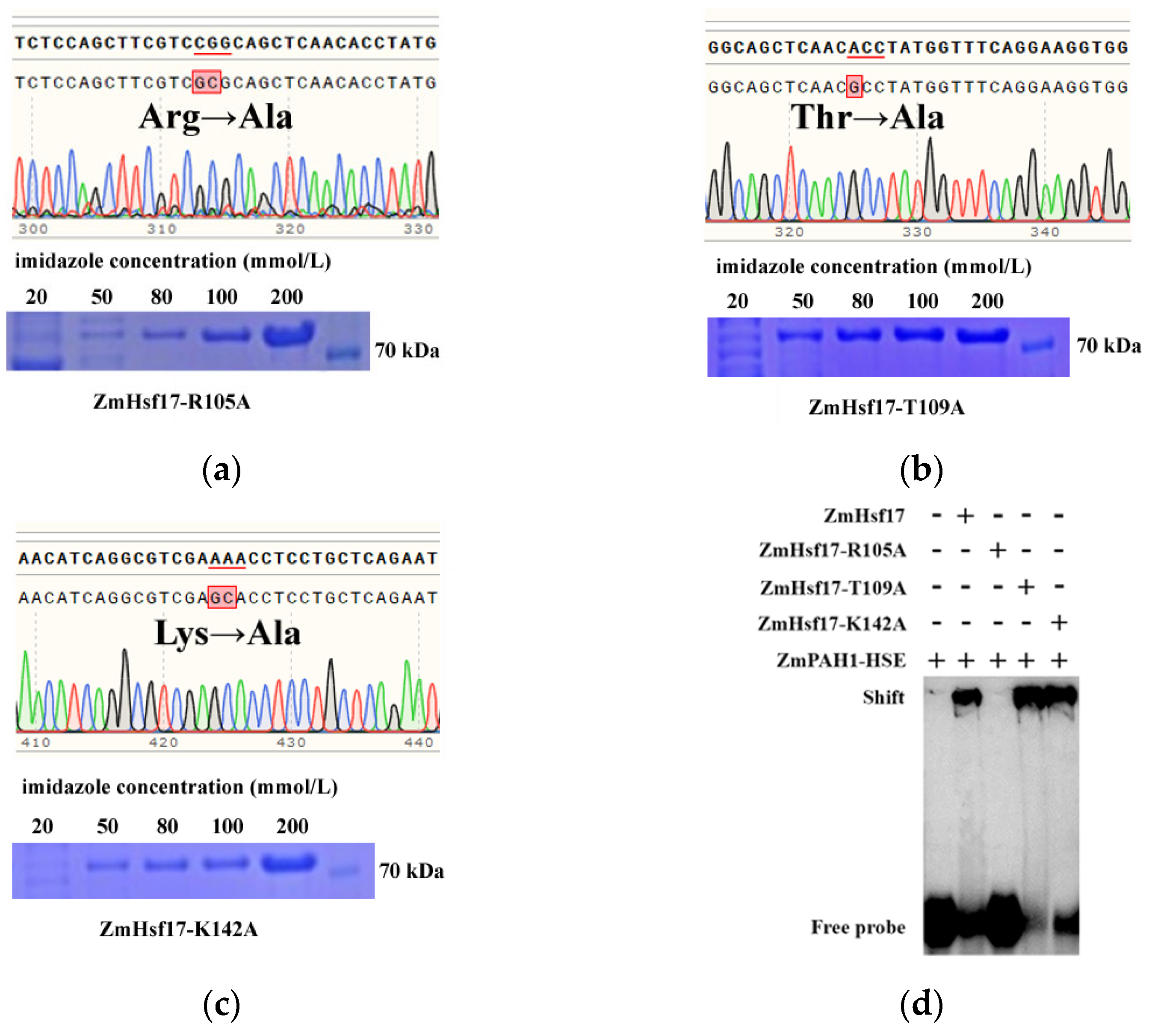
2.2. R105A Substitution Impairs DBD-HSE Binding
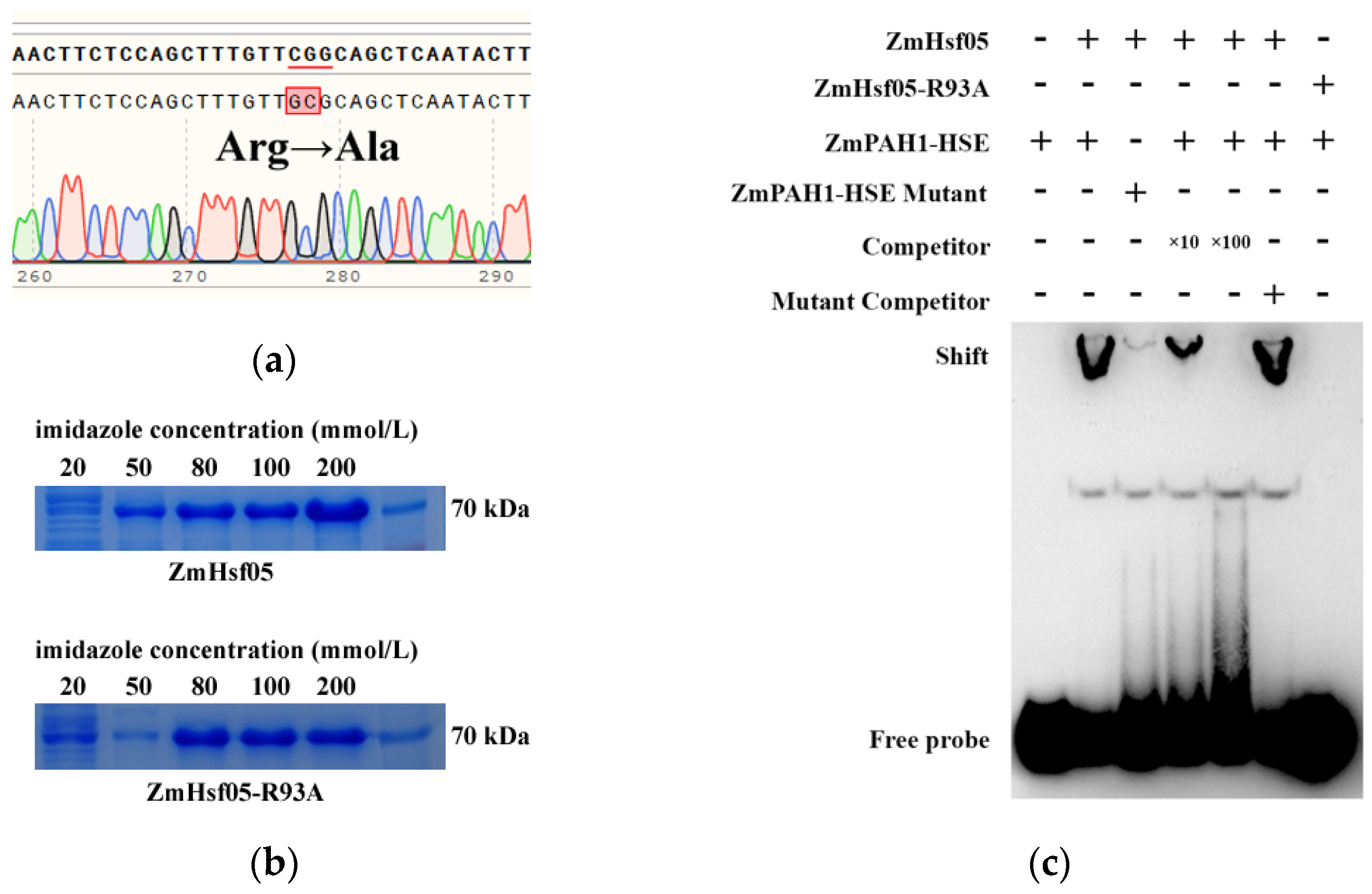
2.3. Conserved Arginine Function Validated in the Paralog ZmHsf05
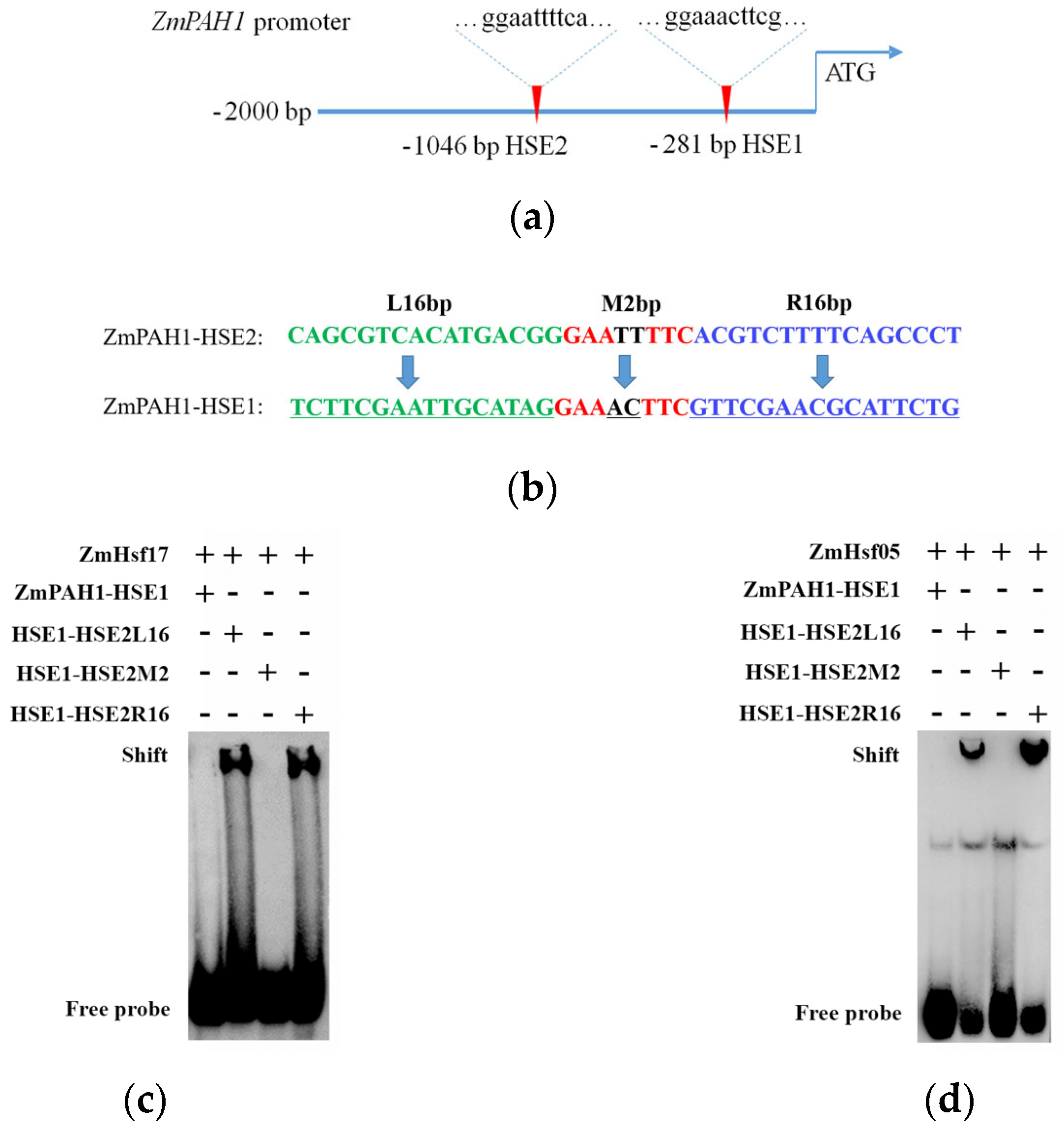
2.4. Flanking Sequences Affect HSE Recognition Specificity
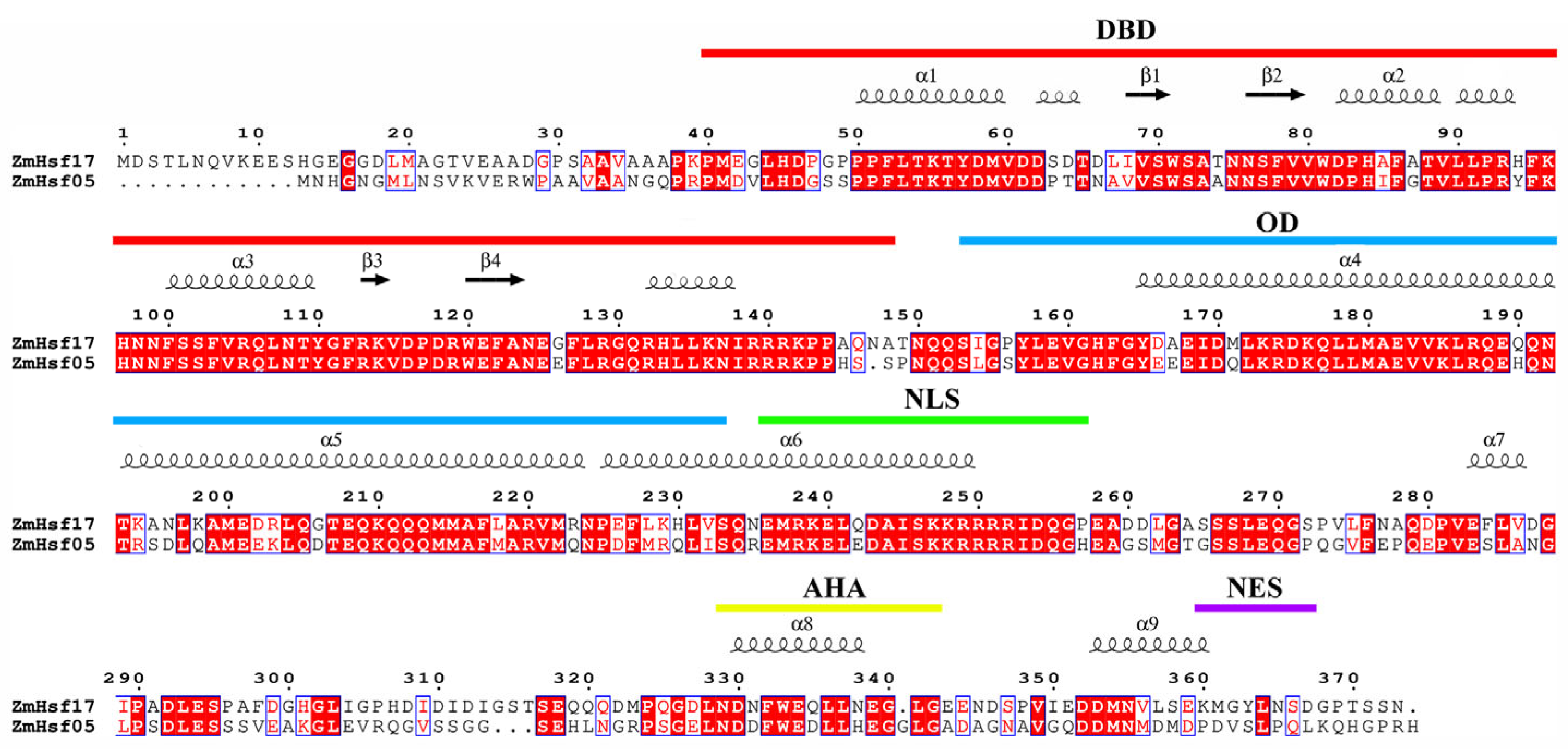
2.5. Evolutionary Analysis of Hsf-DBD
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Gene Cloning
4.2. Protein–DNA Docking via AlphaFold 3
4.3. Site-Directed Mutagenesis by Overlap Extension PCR
4.4. Recombinant Protein Expression and Purification
4.5. Electrophoretic Mobility Shift Assay (EMSA)
4.6. Phylogenetic Tree of Hsf-DBD Regions in 25 Species
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nawaz, M.; Sun, J.; Shabbir, S.; Khattak, W.A.; Ren, G.; Nie, X.; Bo, Y.; Javed, Q.; Du, D.; Sonne, C. A review of plants strategies to resist biotic and abiotic environmental stressors. Sci. Total Environ. 2023, 900, 165832. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Shahzad, B.; Kumar, V.; Kohli, S.K.; Sidhu, G.P.; Bali, A.S.; Handa, N.; Kapoor, D.; Bhardwaj, R.; Zheng, B. Phytohormones regulate accumulation of osmolytes under abiotic stress. Biomolecules 2019, 9, 285. [Google Scholar] [CrossRef] [PubMed]
- Scharf, K.D.; Berberich, T.; Ebersberger, I.; Nover, L. The plant heat stress transcription factor (Hsf) family: Structure, function and evolution. Biochim. Biophys. Acta 2012, 1819, 104–119. [Google Scholar] [CrossRef]
- Si, W.; Liang, Q.; Chen, L.; Song, F.; Chen, Y.; Jiang, H. Ectopic overexpression of Maize heat stress transcription factor ZmHsf05 confers drought tolerance in transgenic rice. Genes 2021, 12, 1568. [Google Scholar] [CrossRef]
- Neudegger, T.; Verghese, J.; Hayer-Hartl, M.; Hartl, F.U.; Bracher, A. Structure of human heat-shock transcription factor 1 in complex with DNA. Nat. Struct. Mol. Biol. 2016, 23, 140–146. [Google Scholar] [CrossRef]
- Dorantes-Palma, D.; Pérez-Mora, S.; Azuara-Liceaga, E.; Pérez-Rueda, E.; Pérez-Ishiwara, D.G.; Coca-González, M.; Medel-Flores, M.O.; Gómez-García, C. Screening and structural characterization of heat shock response elements (HSEs) in entamoeba histolytica promoters. Int. J. Mol. Sci. 2024, 25, 1319. [Google Scholar] [CrossRef]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef]
- Scharf, K.D.; Rose, S.; Zott, W.; Schöff, F.; Nover, L. Three tomato genes code for heat stress transcription factors with a region of remarkable homology to the DNA-binding domain of the yeast HSF. EMBO J. 1990, 9, 4495–4501. [Google Scholar] [CrossRef]
- Guo, M.; Liu, J.H.; Ma, X.; Luo, D.X.; Gong, Z.H.; Lu, M.H. The Plant Heat stress transcription factors (HSFs): Structure, regulation, and function in response to abiotic stresses. Front. Plant Sci. 2016, 7, 114. [Google Scholar] [CrossRef]
- Li, Z.; Howell, S. Heat stress responses and thermotolerance in maize. Int. J. Mol. Sci. 2021, 22, 948. [Google Scholar] [CrossRef]
- Nover, L.; Bharti, K.; Döring, P.; Mishra, S.K.; Ganguli, A.; Scharf, K.D. Arabidopsis and the heat stress transcription factor world: How many heat stress transcription factors do we need. Cell Stress Chaperones 2001, 6, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Sorger, P.K.; Pelham, H.R. Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell 1988, 54, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Liu, H.T.; Sun, D.Y.; Zhou, R.G. Ca2+ and calmodulin modulate DNA-binding activity of maize heat shock transcription factor in vitro. Plant Cell Physiol. 2004, 45, 627–634. [Google Scholar] [CrossRef]
- Feng, H.; Wang, S.; Guo, L.; Punekar, A.S.; Ladenstein, R.; Wang, D.C.; Liu, W. MD simulation of high-resolution X-ray structures reveals post-translational modification dependent conformational changes in HSF-DNA interaction. Protein Cell 2016, 7, 916–920. [Google Scholar] [CrossRef]
- Xiao, Z.; Guo, L.; Zhang, Y.; Cui, L.; Dai, Y.; Lan, Z.; Zhang, Q.; Wang, S.; Liu, W. Structural analysis of missense mutations occurring in the DNA-binding domain of HSF4 associated with congenital cataracts. J. Struct. Biol. X. 2020, 4, 100015. [Google Scholar] [CrossRef]
- Baniwal, S.K.; Bharti, K.; Chan, K.Y.; Fauth, M.; Ganguli, A.; Kotak, S.; Mishra, S.K.; Nover, L.; Port, M.; Scharf, K.D.; et al. Heat stress response in plants: A complex game with chaperones and more than twenty heat stress transcription factors. J. Biosci. 2004, 29, 471–487. [Google Scholar] [CrossRef]
- Jaeger, A.M.; Makley, L.N.; Gestwicki, J.E.; Thiele, D.J. Genomic Heat shock element sequences drive cooperative human heat shock factor 1 DNA binding and selectivity. J. Biol. Chem. 2014, 289, 30459–30469. [Google Scholar] [CrossRef]
- Jung, H.S.; Crisp, P.A.; Estavillo, G.M.; Cole, B.; Hong, F.; Mockler, T.C.; Pogson, B.J.; Chory, J. Subset of heat-shock transcription factors required for the early response of Arabidopsis to excess light. Proc. Natl. Acad. Sci. USA 2013, 110, 14474–14479. [Google Scholar] [CrossRef]
- Rehman, A.; Atif, R.M.; Azhar, M.T.; Peng, Z.; Li, H.; Qin, G.; Jia, Y.; Pan, Z.; He, S.; Qayyum, A.; et al. Genome wide identification, classification and functional characterization of heat shock transcription factors in cultivated and ancestral cottons (Gossypium spp.). Int. J. Biol. Macromol. 2021, 182, 1507–1527. [Google Scholar] [CrossRef]
- Zhang, H.; Meng, X.; Liu, R.; Li, R.; Wang, Y.; Ma, Z.; Liu, Z.; Duan, S.; Li, G.; Guo, X. Heat shock factor ZmHsf17 positively regulates phosphatidic acid phosphohydrolase ZmPAH1 and enhances maize thermotolerance. J. Exp. Bot. 2025, 76, 493–512. [Google Scholar] [CrossRef]
- Zhang, H.; Meng, X.; Li, R.; Ma, Z.; Liu, R.; Liu, Z.; Duan, S.; Zhang, W.; Li, G.; Guo, X. Intron retention via alternative splicing affects the thermotolerance regulation of ZmHsf17. Physiol. Plant. 2024, 176, e14138. [Google Scholar] [CrossRef]
- Zhang, H.; Li, G.; Fu, C.; Duan, S.; Hu, D.; Guo, X. Genome-wide identification, transcriptome analysis and alternative splicing events of Hsf family genes in maize. Sci. Rep. 2020, 10, 8073. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Enoki, Y. Novel aspects of heat shock factors: DNA recognition, chromatin modulation and gene expression. FEBS J. 2010, 277, 4140–4149. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Ohama, N.; Nakajima, J.; Kidokoro, S.; Mizoi, J.; Nakashima, K.; Maruyama, K.; Kim, J.M.; Seki, M.; Todaka, D.; et al. Arabidopsis HsfA1 transcription factors function as the main positive regulators in heat shock-responsive gene expression. Mol. Genet. Genom. 2011, 286, 321–332. [Google Scholar] [CrossRef]
- Jaeger, A.M.; Pemble, C.W.; Sistonen, L.; Thiele, D.J. Structures of HSF2 reveal mechanisms for differential regulation of human heat-shock factors. Nat. Struct. Mol. Biol. 2016, 23, 147–154. [Google Scholar] [CrossRef]
- Kotak, S.; Port, M.; Ganguli, A.; Bicker, F.; von Koskull-Döring, P. Characterization of C-terminal domains of Arabidopsis heat stress transcription factors (Hsfs) and identification of a new signature combination of plant class A Hsfs with AHA and NES motifs essential for activator function and intracellular localization. Plant J. 2004, 39, 98–112. [Google Scholar] [CrossRef]
- Vuister, G.W.; Kim, S.J.; Orosz, A.; Marquardt, J.; Wu, C.; Bax, A. Solution structure of the DNA-binding domain of Drosophila heat shock transcription factor. Nat. Struct. Biol. 1994, 1, 605–614. [Google Scholar] [CrossRef]
- Feng, N.; Feng, H.; Wang, S.; Punekar, A.S.; Ladenstein, R.; Wang, D.C.; Zhang, Q.; Ding, J.; Liu, W. Structures of heat shock factor trimers bound to DNA. iScience 2021, 24, 102951. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, L.; Yu, F.; Liu, Y.; Liang, Q.; Deng, G.; Chen, G.; Liu, M.; Xiao, X. HSF1 is a transcriptional activator of IL-10 gene expression in RAW264.7 macrophages. Inflammation 2012, 35, 1558–1566. [Google Scholar] [CrossRef]
- Guertin, M.J.; Lis, J.T. Chromatin landscape dictates HSF binding to target DNA elements. PLoS Genet. 2010, 6, e1001114. [Google Scholar] [CrossRef]
- Lou, H.; Li, S.; Shi, Z.; Zou, Y.; Zhang, Y.; Huang, X.; Yang, D.; Yang, Y.; Li, Z.; Xu, C. Engineering source-sink relations by prime editing confers heat-stress resilience in tomato and rice. Cell 2025, 188, 530–549. [Google Scholar] [CrossRef]
- El-Shershaby, A.; Ullrich, S.; Simm, S.; Scharf, K.D.; Schleiff, E.; Fragkostefanakis, S. Functional diversification of tomato HsfA1 factors is based on DNA binding domain properties. Gene 2019, 714, 143985. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Ma, Z.; Li, G.; Meng, X.; Duan, S.; Liu, Z.; Zhao, M.; Guo, X.; Zhang, H. The Structural Deciphering of the α3 Helix Within ZmHsfA2’S DNA-Binding Domain for the Recognition of Heat Shock Elements in Maize. Plants 2025, 14, 1950. https://doi.org/10.3390/plants14131950
Wang Y, Ma Z, Li G, Meng X, Duan S, Liu Z, Zhao M, Guo X, Zhang H. The Structural Deciphering of the α3 Helix Within ZmHsfA2’S DNA-Binding Domain for the Recognition of Heat Shock Elements in Maize. Plants. 2025; 14(13):1950. https://doi.org/10.3390/plants14131950
Chicago/Turabian StyleWang, Yantao, Zhenyu Ma, Guoliang Li, Xiangzhao Meng, Shuonan Duan, Zihui Liu, Min Zhao, Xiulin Guo, and Huaning Zhang. 2025. "The Structural Deciphering of the α3 Helix Within ZmHsfA2’S DNA-Binding Domain for the Recognition of Heat Shock Elements in Maize" Plants 14, no. 13: 1950. https://doi.org/10.3390/plants14131950
APA StyleWang, Y., Ma, Z., Li, G., Meng, X., Duan, S., Liu, Z., Zhao, M., Guo, X., & Zhang, H. (2025). The Structural Deciphering of the α3 Helix Within ZmHsfA2’S DNA-Binding Domain for the Recognition of Heat Shock Elements in Maize. Plants, 14(13), 1950. https://doi.org/10.3390/plants14131950