
The Tissue Expression Divergence of the WUSCHEL-Related Homeobox Gene Family in the Evolution of Nelumbo

Abstract
1. Introduction
2. Results
2.1. Identification of WOXs in N. lutea
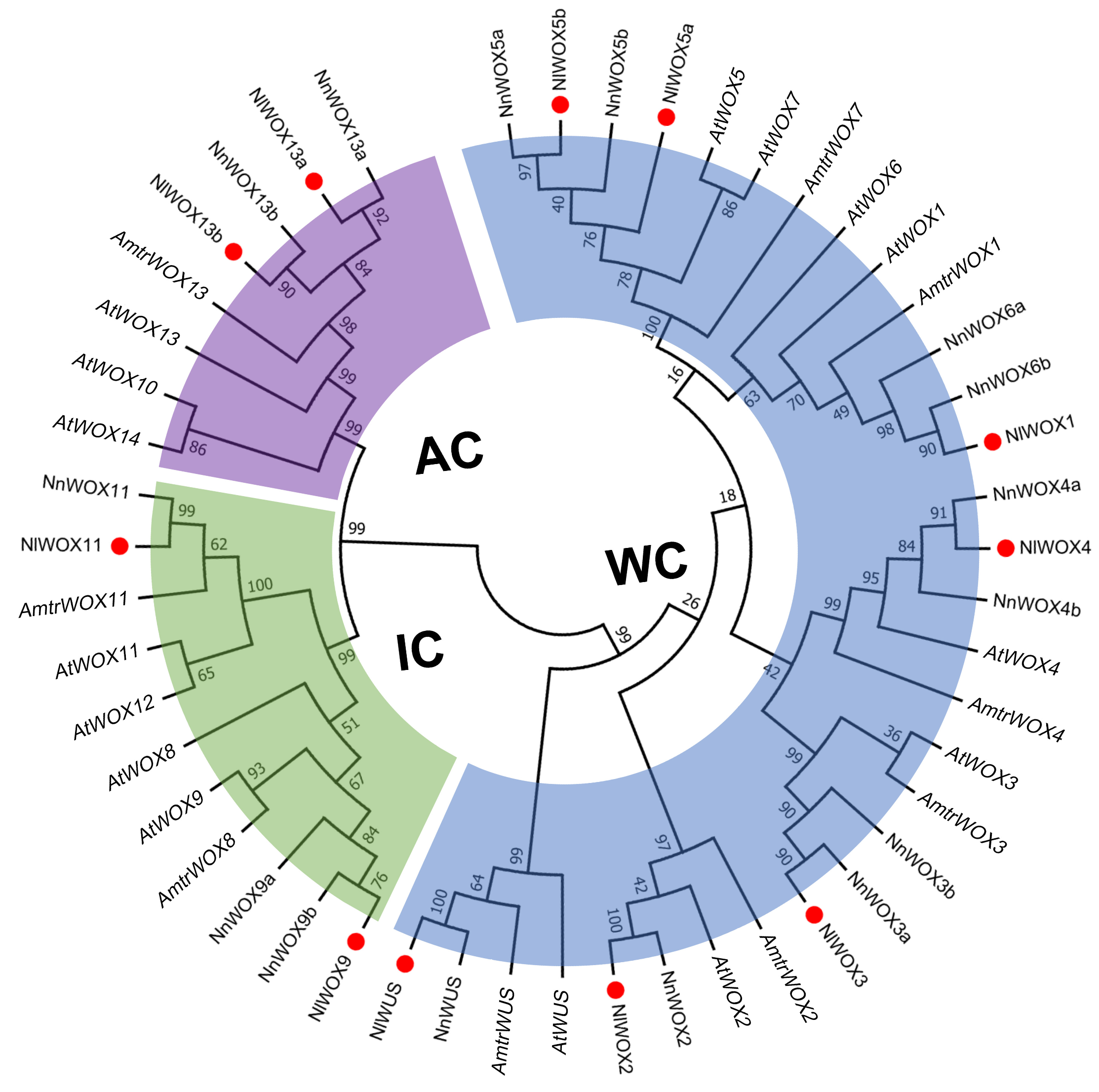
2.2. Phylogenetic Tree of NlWOX Proteins
2.3. Gene Duplication and Synteny Analysis
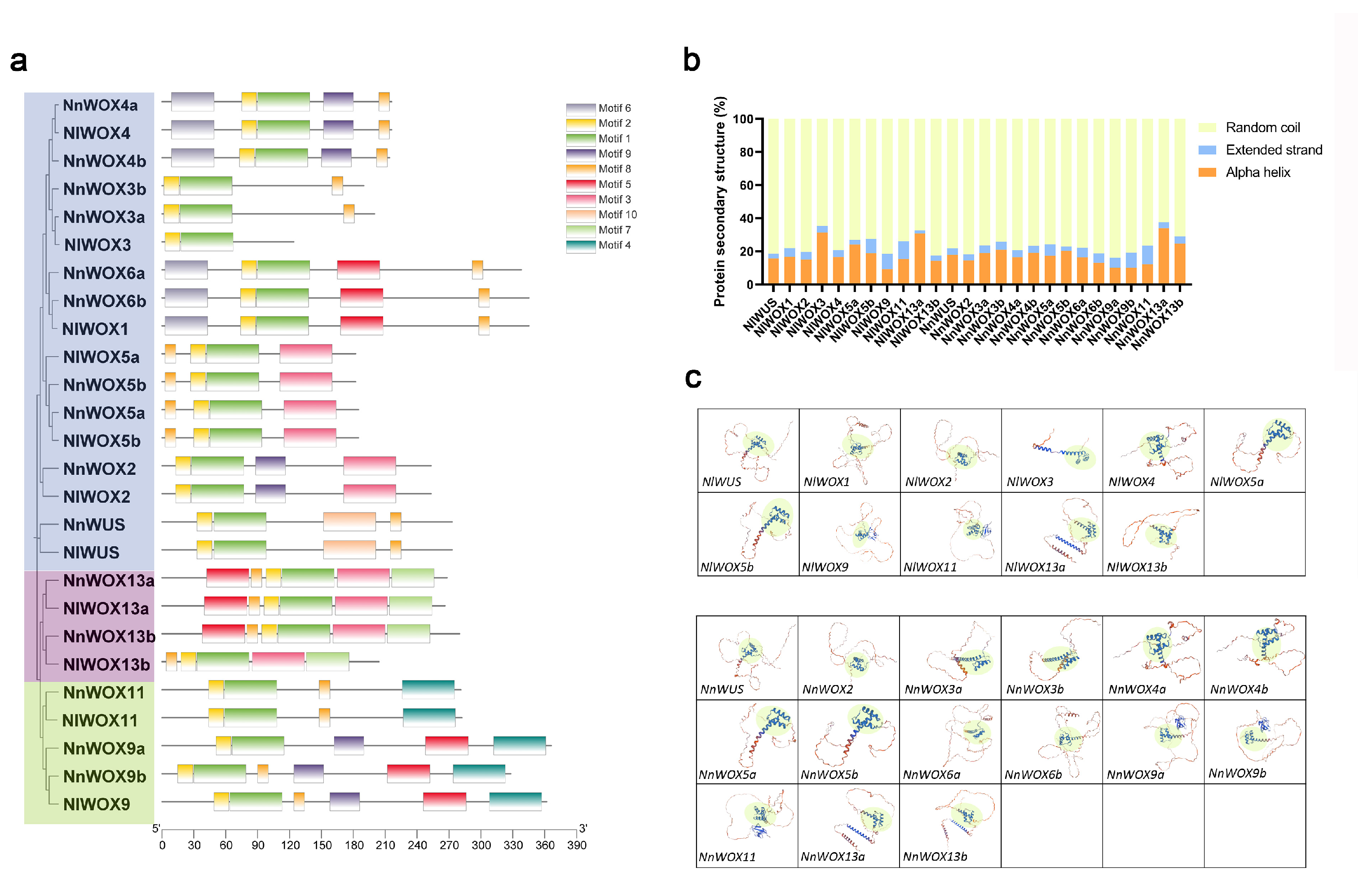
2.4. Conserved Motifs and Protein Conformation of WOX Genes in Nelumbo
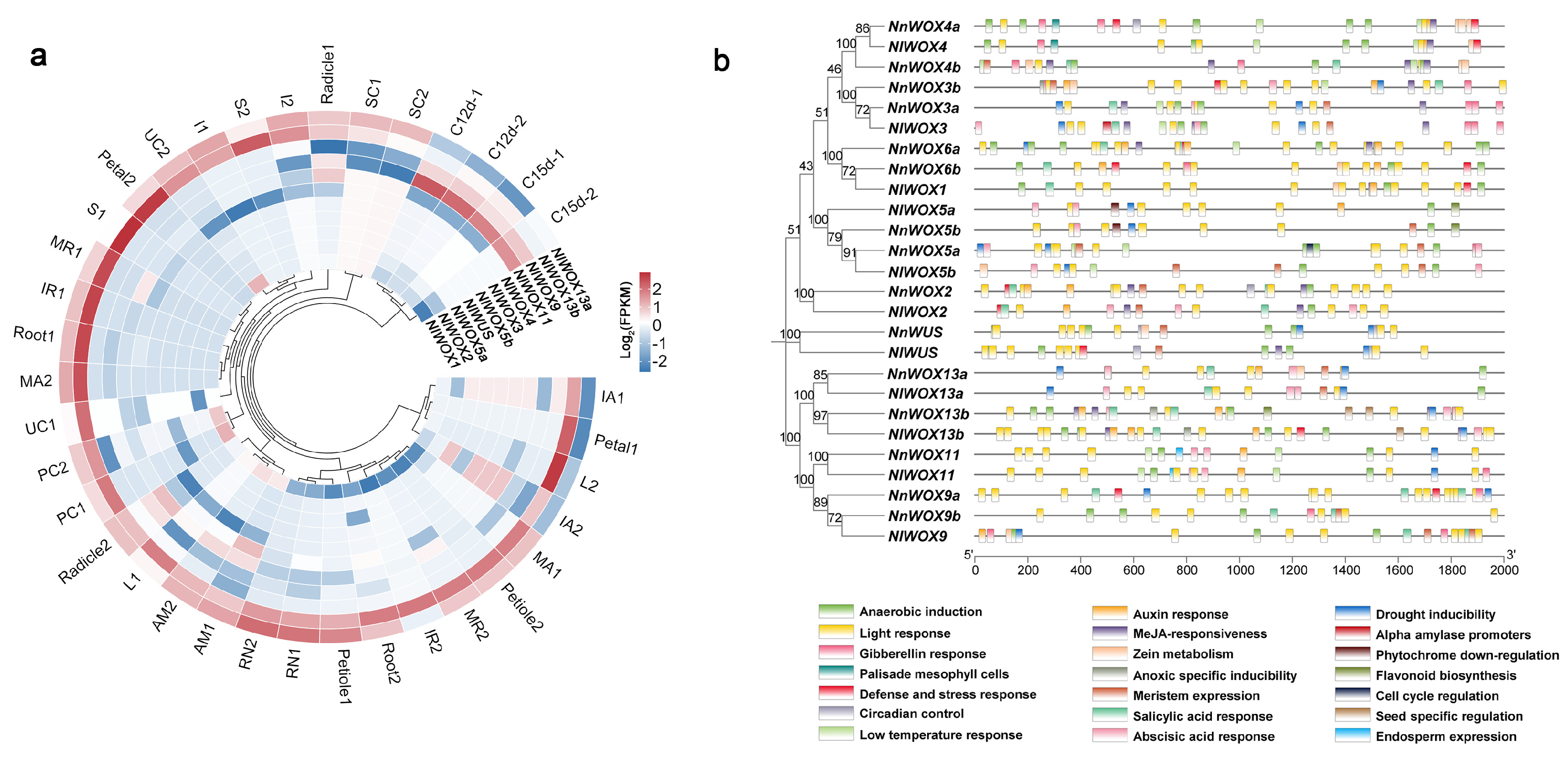
2.5. Tissue Expression Pattern Divergence of WOX Proteins in Lotuses
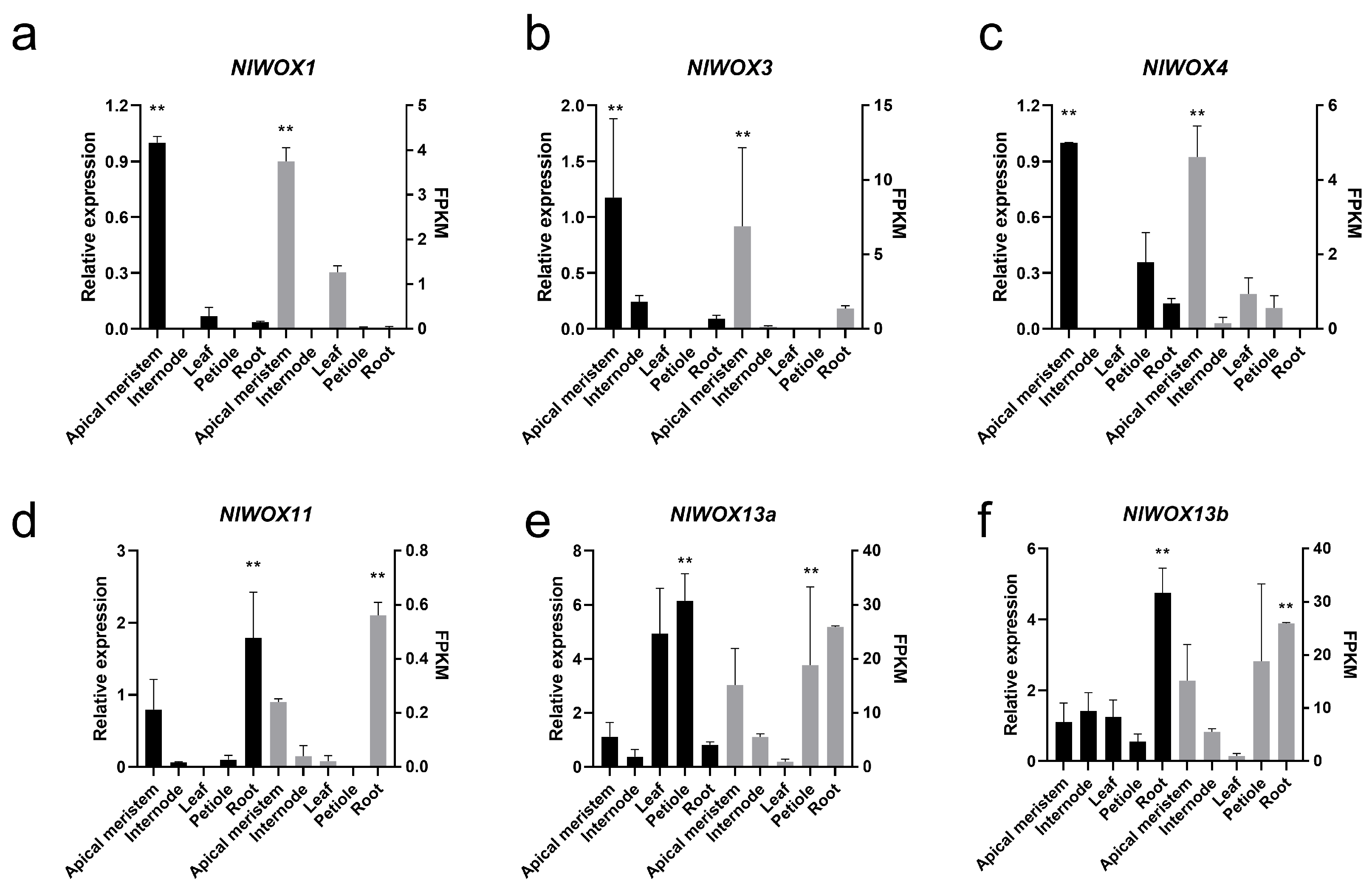
2.6. qRT-PCR Experiments of NlWOX Genes
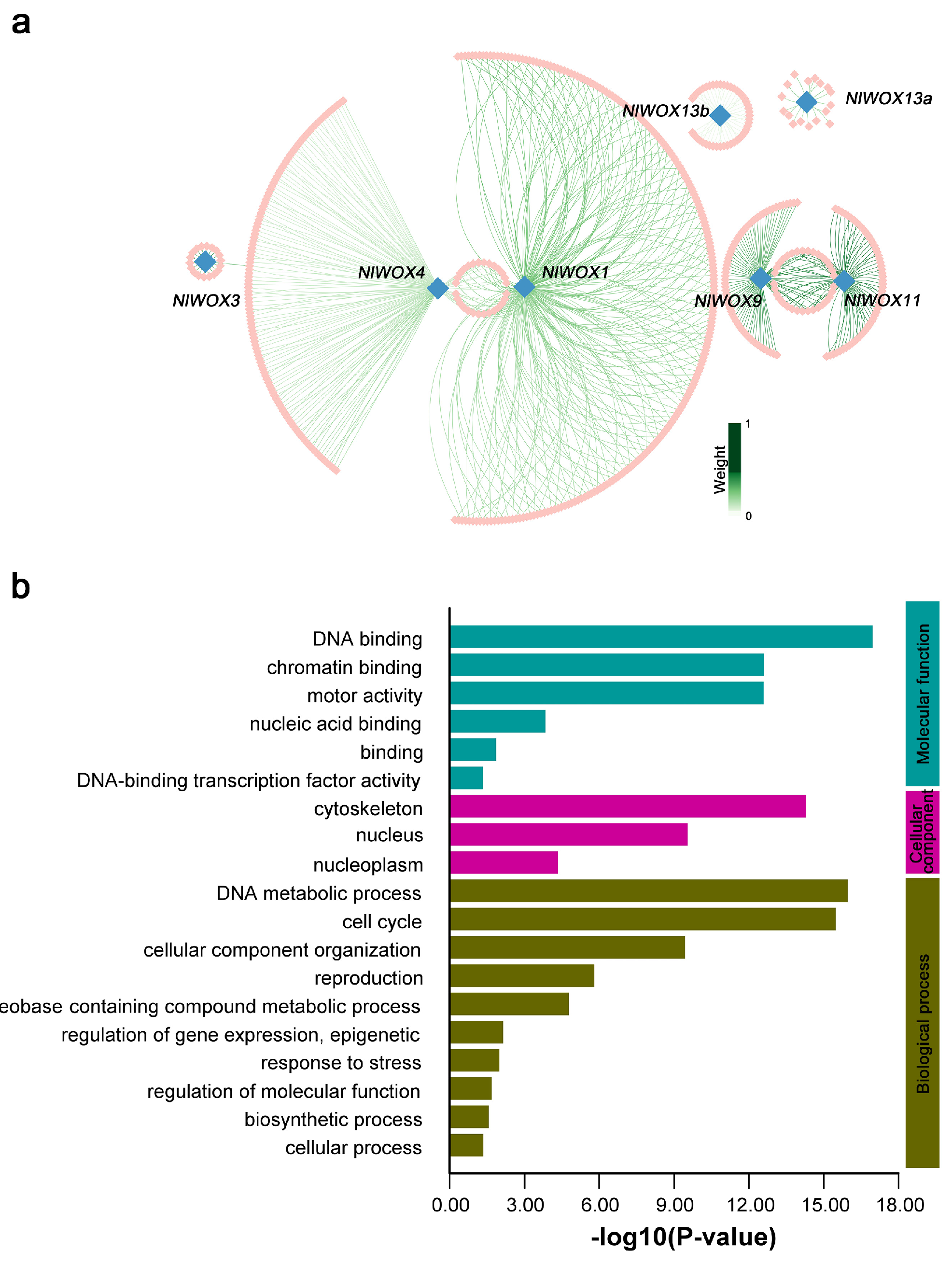
2.7. Co-Expressed Relationships of NlWOX Genes
3. Discussion
4. Materials and Methods
4.1. Identification of WOX Genes in Nelumbo lutea
4.2. Chromosome Location and Physicochemical Characteristics of NlWOX Genes
4.3. Phylogenetic Analysis of NlWOXs
4.4. Interspecies and Intraspecies Synteny Analyses
4.5. GO Enrichment Analysis
4.6. Tissue Expression Profiles of NlWOXs
4.7. Quantitative Real-Time PCR Experiments
4.8. Comparative Co-Expression Gene Network Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feng, B.H.; Han, Y.C.; Xiao, Y.Y.; Kuang, J.F.; Fan, Z.Q.; Chen, J.Y.; Lu, W.J. The banana fruit Dof transcription factor MaDof23 acts as a repressor and interacts with MaERF9 in regulating ripening-related genes. J. Exp. Bot. 2016, 67, 2263–2275. [Google Scholar] [CrossRef] [PubMed]
- Miyashima, S.; Roszak, P.; Sevilem, I.; Toyokura, K.; Blob, B.; Heo, J.O.; Mellor, N.; Help-Rinta-Rahko, H.; Otero, S.; Smet, W.; et al. Mobile PEAR transcription factors integrate positional cues to prime cambial growth. Nature 2019, 565, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.A.; Jakoby, M.; Werber, M.; Martin, C.; Weisshaar, B.; Bailey, P.C. The basic helix-loop-helix transcription factor family in plants: A genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 2003, 20, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Rensing, S.A.; Lang, D.; Zimmer, A.D.; Terry, A.; Salamov, A.; Shapiro, H.; Nishiyama, T.; Perroud, P.F.; Lindquist, E.A.; Kamisugi, Y.; et al. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 2008, 319, 64–69. [Google Scholar] [CrossRef]
- Qiu, Y.; Li, Z.; Köhler, C. Ancestral duplication of MADS-box genes in land plants empowered the functional divergence between sporophytes and gametophytes. New Phytol. 2024, 244, 358–363. [Google Scholar] [CrossRef]
- Wheeler, L.C.; Walker, J.F.; Ng, J.; Deanna, R.; Dunbar-Wallis, A.; Backes, A.; Pezzi, P.H.; Palchetti, M.V.; Robertson, H.M.; Monaghan, A.; et al. Transcription factors evolve faster than their structural gene targets in the flavonoid pigment pathway. Mol. Biol. Evol. 2022, 39, 3. [Google Scholar] [CrossRef]
- Zou, X.; Sun, H. DOF transcription factors: Specific regulators of plant biological processes. Front. Plant Sci. 2023, 14, 1044918. [Google Scholar] [CrossRef]
- Delaux, P.M.; Hetherington, A.J.; Coudert, Y.; Delwiche, C.; Dunand, C.; Gould, S.; Kenrick, P.; Li, F.W.; Philippe, H.; Rensing, S.A.; et al. Reconstructing trait evolution in plant evo-devo studies. Curr. Biol. 2019, 29, R1110–R1118. [Google Scholar] [CrossRef]
- Ruprecht, C.; Mutwil, M.; Saxe, F.; Eder, M.; Nikoloski, Z.; Persson, S. Large-scale co-expression approach to dissect secondary cell wall formation across plant species. Front. Plant Sci. 2011, 2, 23. [Google Scholar] [CrossRef]
- Tzfadia, O.; Amar, D.; Bradbury, L.M.; Wurtzel, E.T.; Shamir, R. The MORPH algorithm: Ranking candidate genes for membership in Arabidopsis and tomato pathways. Plant Cell 2012, 24, 4389–4406. [Google Scholar] [CrossRef]
- Kamiya, N.; Nagasaki, H.; Morikami, A.; Sato, Y.; Matsuoka, M. Isolation and characterization of a rice WUSCHEL-type homeobox gene that is specifically expressed in the central cells of a quiescent center in the root apical meristem. Plant J. 2003, 35, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.F.; Schoof, H.; Haecker, A.; Lenhard, M.; Jürgens, G.; Laux, T. Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell 1998, 95, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, R.; Ji, J.; Kelsey, E.; Ohtsu, K.; Schnable, P.S.; Scanlon, M.J. Tissue specificity and evolution of meristematic WOX3 function. Plant Physiol. 2009, 149, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Park, S.O.; Zheng, Z.; Oppenheimer, D.G.; Hauser, B.A. The PRETTY FEW SEEDS2 gene encodes an Arabidopsis homeodomain protein that regulates ovule development. Development 2005, 132, 841–849. [Google Scholar] [CrossRef]
- Deveaux, Y.; Toffano-Nioche, C.; Claisse, G.; Thareau, V.; Morin, H.; Laufs, P.; Moreau, H.; Kreis, M.; Lecharny, A. Genes of the most conserved WOX clade in plants affect root and flower development in Arabidopsis. BMC Evol. Biol. 2008, 8, 291. [Google Scholar] [CrossRef]
- van der Graaff, E.; Laux, T.; Rensing, S.A. The WUS homeobox-containing (WOX) protein family. Genome Biol. 2009, 10, 248. [Google Scholar] [CrossRef]
- Lin, H.; Niu, L.; McHale, N.A.; Ohme-Takagi, M.; Mysore, K.S.; Tadege, M. Evolutionarily conserved repressive activity of WOX proteins mediates leaf blade outgrowth and floral organ development in plants. Proc. Natl. Acad. Sci. USA 2013, 110, 366–371. [Google Scholar] [CrossRef]
- Shafique Khan, F.; Zeng, R.F.; Gan, Z.M.; Zhang, J.Z.; Hu, C.G. Genome-wide identification and expression profiling of the WOX gene family in citrus sinensis and functional analysis of a CsWUS member. Int. J. Mol. Sci. 2021, 22, 4919. [Google Scholar] [CrossRef]
- Lv, J.; Feng, Y.; Jiang, L.; Zhang, G.; Wu, T.; Zhang, X.; Xu, X.; Wang, Y.; Han, Z. Genome-wide identification of WOX family members in nine Rosaceae species and a functional analysis of MdWOX13-1 in drought resistance. Plant Sci. 2023, 328, 111564. [Google Scholar] [CrossRef]
- Zhao, Y.; Cheng, S.; Song, Y.; Huang, Y.; Zhou, S.; Liu, X.; Zhou, D.X. The Interaction between rice ERF3 and WOX11 promotes crown root development by regulating gene expression involved in cytokinin signaling. Plant Cell 2015, 27, 2469–2483. [Google Scholar] [CrossRef]
- Batcho, A.A.; Nwogwugwu, J.O.; Ali, M.; Jabbar, B.; Javaid, A.; Fellner, M. Identification and characterisation of blue light photoreceptor gene family and their expression in tomato (Solanum lycopersicum) under cold stress. Funct. Plant Biol. 2022, 49, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Feng, H.; Hu, Q.; Qu, H.; Chen, A.; Yu, L.; Xu, G. Improving rice tolerance to potassium deficiency by enhancing OsHAK16p:WOX11-controlled root development. Plant Biotechnol. J. 2015, 13, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Cheng, X.; Lan, T.; Guo, X.; Su, Z.; An, X.; Zheng, Y.; Cui, H.; Wu, W.; Lan, T. OsSPL10 controls trichome development by interacting with OsWOX3B at both transcription and protein levels in rice (Oryza sativa L.). Crop J. 2023, 11, 1711–1718. [Google Scholar] [CrossRef]
- Rasheed, H.; Shi, L.; Winarsih, C.; Jakada, B.H.; Chai, R.; Huang, H. Plant growth regulators: An overview of WOX gene family. Plants 2024, 13, 3108. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.S.; Goher, F.; Hu, C.G.; Zhang, J.Z. WUSCHEL-related homeobox (WOX) transcription factors: Key regulators in combating abiotic stresses in plants. Hortic. Adv. 2024, 2, 2. [Google Scholar] [CrossRef]
- Wang, L.Q.; Wen, S.S.; Wang, R.; Wang, C.; Gao, B.; Lu, M.Z. PagWOX11/12a activates PagCYP736A12 gene that facilitates salt tolerance in poplar. Plant Biotechnol. J. 2021, 19, 2249–2260. [Google Scholar] [CrossRef]
- Minh-Thu, P.T.; Kim, J.S.; Chae, S.; Jun, K.M.; Lee, G.S.; Kim, D.E.; Cheong, J.J.; Song, S.I.; Nahm, B.H.; Kim, Y.K. A WUSCHEL homeobox transcription factor, OsWOX13, enhances drought tolerance and triggers early flowering in rice. Mol. Cells 2018, 41, 781–798. [Google Scholar]
- Stuart, J.M.; Segal, E.; Koller, D.; Kim, S.K. A gene-coexpression network for global discovery of conserved genetic modules. Science 2003, 302, 249–255. [Google Scholar] [CrossRef]
- Tai, Y.; Liu, C.; Yu, S.; Yang, H.; Sun, J.; Guo, C.; Huang, B.; Liu, Z.; Yuan, Y.; Xia, E.; et al. Gene co-expression network analysis reveals coordinated regulation of three characteristic secondary biosynthetic pathways in tea plant (Camellia sinensis). BMC Genom. 2018, 19, 616. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Deng, X.; Zhang, M.; Sun, H.; Gao, L.; Song, H.; Xin, J.; Ming, R.; Yang, D.; et al. Transcription factor NnMYB5 controls petal color by regulating GLUTATHIONE S-TRANSFERASE2 in Nelumbo nucifera. Plant Physiol. 2023, 193, 1213–1226. [Google Scholar] [CrossRef]
- Shi, T.; Rahmani, R.S.; Gugger, P.F.; Wang, M.; Li, H.; Zhang, Y.; Li, Z.; Wang, Q.; Van de Peer, Y.; Marchal, K.; et al. Distinct expression and methylation patterns for genes with different fates following a single whole-genome duplication in flowering plants. Mol. Biol. Evol. 2020, 37, 2394–2413. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yang, M.; Li, L.; Li, H.; Yang, D.; Shi, T.; Yang, P. Whole genome re-sequencing reveals evolutionary patterns of sacred lotus (Nelumbo nucifera). J. Integr. Plant Biol. 2018, 60, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, X.; Zhang, Y.; Gao, Z.; Liang, Y.; Chen, J.; Shi, T. Nelumbo genome database, an integrative resource for gene expression and variants of Nelumbo nucifera. Sci. Data 2021, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Qiu, X.Y.; Dai, Y.J.; Nyonga, T.M.; Li, C.C. Genome-wide identification and co-expression networks of WOX gene family in Nelumbo nucifera. Plants 2024, 13, 720. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Van de Peer, Y.; Chen, J.; Marchal, K.; Shi, T. Evolution of isoform-level gene expression patterns across tissues during lotus species divergence. Plant J. 2022, 112, 830–846. [Google Scholar] [CrossRef]
- Zheng, P.; Sun, H.; Liu, J.; Lin, J.; Zhang, X.; Qin, Y.; Zhang, W.; Xu, X.; Deng, X.; Yang, D.; et al. Comparative analyses of American and Asian lotus genomes reveal insights into petal color, carpel thermogenesis and domestication. Plant J. 2022, 110, 1498–1515. [Google Scholar] [CrossRef]
- Lou, X.; Wang, J.; Wang, G.; He, D.; Shang, W.; Song, Y.; Wang, Z.; He, S. Genome-wide analysis of the WOX family and its expression pattern in root development of Paeonia ostii. Int. J. Mol. Sci. 2024, 25, 7668. [Google Scholar] [CrossRef]
- Ikeda, M.; Mitsuda, N.; Ohme-Takagi, M. Arabidopsis WUSCHEL is a bifunctional transcription factor that acts as a repressor in stem cell regulation and as an activator in floral patterning. Plant Cell 2009, 21, 3493–3505. [Google Scholar] [CrossRef]
- Zhang, X.; Zong, J.; Liu, J.; Yin, J.; Zhang, D. Genome-wide analysis of WOX gene family in rice, sorghum, maize, Arabidopsis and poplar. J. Integr. Plant Biol. 2010, 52, 1016–1026. [Google Scholar] [CrossRef]
- Ren, H.; Shankle, K.; Cho, M.J.; Tjahjadi, M.; Khanday, I.; Sundaresan, V. Synergistic induction of fertilization-independent embryogenesis in rice egg cells by paternal-genome-expressed transcription factors. Nat. Plants 2024, 10, 1892–1899. [Google Scholar] [CrossRef]
- Sun, P.; Yuan, H.; Pan, J.; Wu, Z.; Li, W.; Wang, X.; Kuang, H.; Chen, J. A WOX homolog disrupted by a transposon led to the loss of spines and contributed to the domestication of lettuce. New Phytol. 2024, 242, 2857–2871. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, W.; Yao, Y.; Zhang, L.; Xu, S.; Zhang, Q.; Huang, T. FRUCTOSE INSENSITIVE1 regulates stem cell function in Arabidopsis in response to fructose signalling. J. Exp. Bot. 2023, 74, 3060–3073. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Hao, Y.; Cui, H. The WUSCHEL related homeobox protein WOX7 regulates the sugar response of lateral root development in Arabidopsis thaliana. Mol. Plant 2016, 9, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Hou, Z.; Zhang, W.; Liang, S.; Huangfu, M.; Zhang, J.; Yang, T.; Dong, J.; Che, D. Genome-wide analysis of the WOX gene family and function exploration of RhWOX331 in rose (R. ‘The Fairy’). Front. Plant Sci. 2024, 15, 1461322. [Google Scholar] [CrossRef]
- Itoh, J.; Sato, Y.; Sato, Y.; Hibara, K.; Shimizu-Sato, S.; Kobayashi, H.; Takehisa, H.; Sanguinet, K.A.; Namiki, N.; Nagamura, Y. Genome-wide analysis of spatiotemporal gene expression patterns during early embryogenesis in rice. Development 2016, 143, 1217–1227. [Google Scholar]
- Yang, Z.; Gong, Q.; Qin, W.; Yang, Z.; Cheng, Y.; Lu, L.; Ge, X.; Zhang, C.; Wu, Z.; Li, F. Genome-wide analysis of WOX genes in upland cotton and their expression pattern under different stresses. BMC Plant Biol. 2017, 17, 113. [Google Scholar] [CrossRef]
- Etchells, J.P.; Provost, C.M.; Mishra, L.; Turner, S.R. WOX4 and WOX14 act downstream of the PXY receptor kinase to regulate plant vascular proliferation independently of any role in vascular organisation. Development 2013, 140, 2224–2234. [Google Scholar] [CrossRef]
- Denis, E.; Kbiri, N.; Mary, V.; Claisse, G.; Conde, E.S.N.; Kreis, M.; Deveaux, Y. WOX14 promotes bioactive gibberellin synthesis and vascular cell differentiation in Arabidopsis. Plant J. 2017, 90, 560–572. [Google Scholar] [CrossRef]
- Li, M.; Hameed, I.; Cao, D.; He, D.; Yang, P. Integrated omics analyses identify key pathways involved in petiole rigidity formation in sacred lotus. Int. J. Mol. Sci. 2020, 21, 5087. [Google Scholar] [CrossRef]
- Abubakar, A.S.; Wu, Y.; Chen, F.; Zhu, A.; Chen, P.; Chen, K.; Qiu, X.; Huang, X.; Zhao, H.; Chen, J.; et al. Comprehensive analysis of WUSCEL-related homeobox gene family in ramie (Boehmeria nivea) indicates its potential role in adventitious root development. Biology 2023, 12, 1475. [Google Scholar] [CrossRef]
- Atilgan, A.R.; Atilgan, C. Computational strategies for protein conformational ensemble detection. Curr. Opin. Struct. Biol. 2022, 72, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, L.; Geng, W.; Cheng, R.; Zhang, H.; Zhou, H. Genome-wide prediction and functional analysis of WOX genes in blueberry. BMC Genom. 2024, 25, 434. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.; Regev, A.; Roy, S. Comparative analysis of gene regulatory networks: From network reconstruction to evolution. Annu. Rev. Cell Dev. Biol. 2015, 31, 399–428. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, M.; Iwase, A.; Ito, T.; Tanaka, H.; Favero, D.S.; Kawamura, A.; Sakamoto, S.; Wakazaki, M.; Tameshige, T.; Fujii, H.; et al. Wound-inducible WUSCHEL-RELATED HOMEOBOX 13 is required for callus growth and organ reconnection. Plant Physiol. 2022, 188, 425–441. [Google Scholar] [CrossRef]
- Ogura, N.; Sasagawa, Y.; Ito, T.; Tameshige, T.; Kawai, S.; Sano, M.; Doll, Y.; Iwase, A.; Kawamura, A.; Suzuki, T.; et al. WUSCHEL-RELATED HOMEOBOX 13 suppresses de novo shoot regeneration via cell fate control of pluripotent callus. Sci. Adv. 2023, 9, eadg6983. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. EggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Zhang, H.; Meltzer, P.; Davis, S. RCircos: An R package for Circos 2D track plots. BMC Bioinform. 2013, 14, 244. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Name | Number of Amino Acids | Molecular Weight | Theoretical PI | Instability Index | Aliphatic Index | Grand Average of Hydropathicity |
|---|---|---|---|---|---|---|---|
| NL1g_04069 | NlWOX9 | 362 | 39,945.94 | 7.15 | 53.16 | 67.62 | −0.464 |
| NL1g_04550 | NlWOX4 | 216 | 24,502.73 | 9.46 | 54.77 | 63.61 | −0.946 |
| NL1g_06482 | NlWUS | 273 | 30,043.36 | 6.83 | 63.71 | 54.32 | −0.746 |
| NL2g_10541 | NlWOX5a | 182 | 20,712.29 | 6.91 | 60.57 | 67.47 | −0.795 |
| NL2g_11918 | NlWOX2 | 253 | 27,883.17 | 6.71 | 50.01 | 63.2 | −0.611 |
| NL2g_11942 | NlWOX3 | 124 | 14,769.82 | 10.01 | 81.83 | 65.4 | −0.99 |
| NL2g_12810 | NlWOX5b | 185 | 21,006.71 | 8.7 | 42.95 | 68.49 | −0.657 |
| NL4g_22694 | NlWOX1 | 345 | 39,317.67 | 6.43 | 57.25 | 53.45 | −0.904 |
| NL5g_27723 | NlWOX13a | 266 | 30,660.55 | 6.08 | 58.31 | 67.82 | −0.812 |
| NL5g_28623 | NlWOX11 | 282 | 30,602.15 | 5.42 | 75.04 | 69.79 | −0.291 |
| NL6g_29420 | NlWOX13b | 204 | 22,930.45 | 6.06 | 53.57 | 68.38 | −0.793 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zhang, Y. The Tissue Expression Divergence of the WUSCHEL-Related Homeobox Gene Family in the Evolution of Nelumbo. Plants 2025, 14, 1909. https://doi.org/10.3390/plants14131909
Li J, Zhang Y. The Tissue Expression Divergence of the WUSCHEL-Related Homeobox Gene Family in the Evolution of Nelumbo. Plants. 2025; 14(13):1909. https://doi.org/10.3390/plants14131909
Chicago/Turabian StyleLi, Juanjuan, and Yue Zhang. 2025. "The Tissue Expression Divergence of the WUSCHEL-Related Homeobox Gene Family in the Evolution of Nelumbo" Plants 14, no. 13: 1909. https://doi.org/10.3390/plants14131909
APA StyleLi, J., & Zhang, Y. (2025). The Tissue Expression Divergence of the WUSCHEL-Related Homeobox Gene Family in the Evolution of Nelumbo. Plants, 14(13), 1909. https://doi.org/10.3390/plants14131909