
Exploring the Potential of Phytocannabinoids Against Multidrug-Resistant Bacteria


 , ,
, ,  ,
,  , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results and Discussion
2.1. Screening of Phytocannabinoids Against Multidrug-Resistant Pathogens
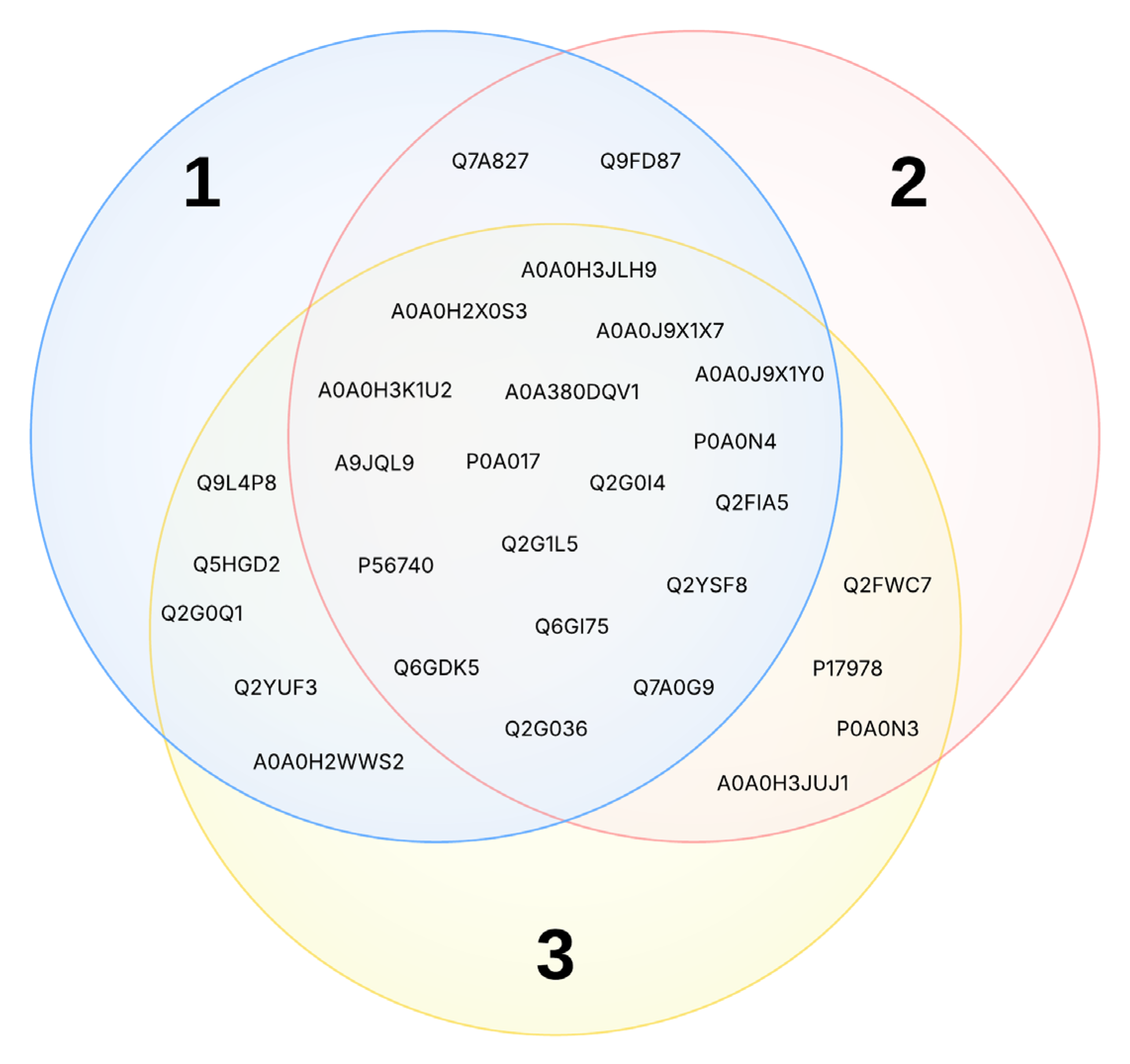
2.2. Inverse Virtual Screening (IVS)
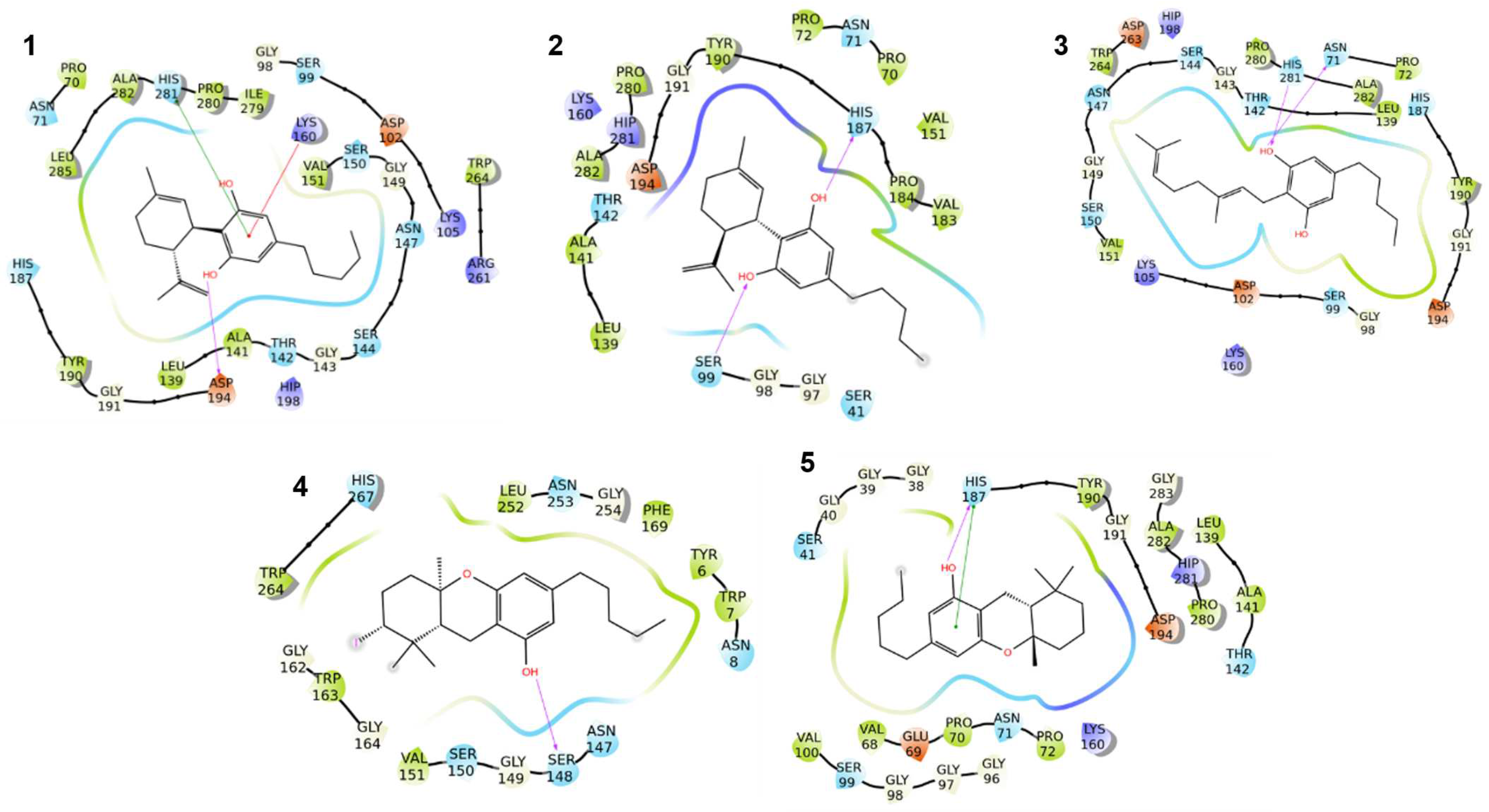
2.2.1. Rationalization of the Results on S. aureus
- a.
- Fatty acid metabolism
- b.
- Nucleotide biosynthesis
- c.
- Folate metabolism
- d.
- Vitamin and cofactor biosynthesis
- e.
- Redox homeostasis
- f.
- Transcriptional regulation
2.2.2. Rationalization of the Results on E. faecium
2.2.3. Rationalization of the Results on B. spizizenii
2.2.4. Implications for Bacterial Multidrug Resistance
3. Materials and Methods
3.1. Phytocannabinoids
3.2. Antibacterial Activity Testing
3.3. Toxicity Assay
3.4. In silico Studies
3.4.1. Ligand and Panel Preparation
3.4.2. Inverse Virtual Screening
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ahmed, S.K.; Hussein, S.; Qurbani, K.; Ibrahim, R.H.; Fareeq, A.; Mahmood, K.A.; Mohamed, M.G. Antimicrobial resistance: Impacts, challenges, and future prospects. J. Med. Surg. Public Health 2024, 2, 100081. [Google Scholar] [CrossRef]
- Dyson, P.J.; Banat, I.M.; Quinn, G.A. War and peace: Exploring microbial defence systems as a source of new antimicrobial therapies. Front. Pharmacol. 2025, 15, 1504901. [Google Scholar] [CrossRef] [PubMed]
- Petrakis, V.; Panopoulou, M.; Rafailidis, P.; Lemonakis, N.; Lazaridis, G.; Terzi, I.; Papazoglou, D.; Panagopoulos, P. The impact of the COVID-19 pandemic on antimicrobial resistance and management of bloodstream infections. Pathogens 2023, 12, 780. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.Y.; Impalli, I.; Poleon, S.; Denoel, P.; Cipriano, M.; Van Boeckel, T.P.; Pecetta, S.; Bloom, D.E.; Nandi, A. Global trends in antibiotic consumption during 2016–2023 and future projections through 2030. Proc. Natl. Acad. Sci. USA 2024, 121, e2411919121. [Google Scholar] [CrossRef]
- Kakkar, A.K.; Shafiq, N.; Singh, G.; Ray, P.; Gautam, V.; Agarwal, R.; Muralidharan, J.; Arora, P. Antimicrobial stewardship programs in resource constrained environments: Understanding and addressing the need of the systems. Front. Public Health 2020, 8, 140. [Google Scholar] [CrossRef]
- Angelini, P. Plant-derived antimicrobials and their crucial role in combating antimicrobial resistance. Antibiotics 2024, 13, 746. [Google Scholar] [CrossRef]
- Appendino, G.; Gibbons, S.; Giana, A.; Pagani, A.; Grassi, G.; Stavri, M.; Smith, E.; Rahman, M.M. Antibacterial cannabinoids from Cannabis sativa: A structure−activity study. J. Nat. Prod. 2008, 71, 1427–1430. [Google Scholar] [CrossRef]
- Cham, P.S.; Deepika; Bhat, R.; Raina, D.; Manhas, D.; Kotwal, P.; Mindala, D.P.; Pandey, N.; Ghosh, A.; Saran, S.; et al. Exploring the antibacterial potential of semisynthetic phytocannabinoid: Tetrahydrocannabidiol (THCBD) as a potential antibacterial agent against sensitive and resistant strains of Staphylococcus aureus. ACS Infect. Dis. 2024, 10, 64–78. [Google Scholar] [CrossRef]
- Luz-Veiga, M.; Amorim, M.; Pinto-Ribeiro, I.; Oliveira, A.L.S.; Silva, S.; Pimentel, L.L.; Rodríguez-Alcalá, L.M.; Madureira, R.; Pintado, M.; Azevedo-Silva, J.; et al. Cannabidiol and cannabigerol exert antimicrobial activity without compromising skin microbiota. Int. J. Mol. Sci. 2023, 24, 2389. [Google Scholar] [CrossRef]
- Farha, M.A.; El-Halfawy, O.M.; Gale, R.T.; MacNair, C.R.; Carfrae, L.A.; Zhang, X.; Jentsch, N.G.; Magolan, J.; Brown, E.D. Uncovering the hidden antibiotic potential of Cannabis. ACS Infect. Dis. 2020, 6, 338–346. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T.; Kavanagh, A.M.; Elliott, A.G.; Zhang, B.; Ramu, S.; Amado, M.; Lowe, G.J.; Hinton, A.O.; Pham, D.M.T.; Zuegg, J.; et al. The antimicrobial potential of cannabidiol. Commun. Biol. 2021, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.; Neira Agonh, D.; Lehmann, C. Antibacterial Effects of Phytocannabinoids. Life 2022, 12, 1394. [Google Scholar] [CrossRef] [PubMed]
- Blebea, N.M.; Pricopie, A.I.; Vlad, R.-A.; Hancu, G. Phytocannabinoids: Exploring Pharmacological Profiles and Their Impact on Therapeutical Use. Int. J. Mol. Sci. 2024, 25, 4204. [Google Scholar] [CrossRef] [PubMed]
- Lauro, G.; Masullo, M.; Piacente, S.; Riccio, R.; Bifulco, G. Inverse Virtual Screening allows the discovery of the biological activity of natural compounds. Biorg. Med. Chem. 2012, 20, 3596–3602. [Google Scholar] [CrossRef]
- Lauro, G.; Romano, A.; Riccio, R.; Bifulco, G. Inverse Virtual Screening of Antitumor Targets: Pilot Study on a Small Database of Natural Bioactive Compounds. J. Nat. Prod. 2011, 74, 1401–1407. [Google Scholar] [CrossRef]
- Ostacolo, C.; Di Sarno, V.; Lauro, G.; Pepe, G.; Musella, S.; Ciaglia, T.; Vestuto, V.; Autore, G.; Bifulco, G.; Marzocco, S.; et al. Identification of an indol-based multi-target kinase inhibitor through phenotype screening and target fishing using inverse virtual screening approach. Eur. J. Med. Chem. 2019, 167, 61–75. [Google Scholar] [CrossRef]
- Scrima, M.; Lauro, G.; Grimaldi, M.; Di Marino, S.; Tosco, A.; Picardi, P.; Gazzerro, P.; Riccio, R.; Novellino, E.; Bifulco, M.; et al. Structural Evidence of N6-Isopentenyladenosine As a New Ligand of Farnesyl Pyrophosphate Synthase. J. Med. Chem. 2014, 57, 7798–7803. [Google Scholar] [CrossRef]
- Chini, M.G.; Lauro, G.; Bifulco, G. Addressing the Target Identification and Accelerating the Repositioning of Anti-Inflammatory/Anti-Cancer Organic Compounds by Computational Approaches. Eur. J. Org. Chem. 2021, 2021, 2966–2981. [Google Scholar] [CrossRef]
- Lopatriello, A.; Caprioglio, D.; Minassi, A.; Schiano Moriello, A.; Formisano, C.; De Petrocellis, L.; Appendino, G.; Taglialatela-Scafati, O. Iodine-mediated cyclization of cannabigerol (CBG) expands the cannabinoid biological and chemical space. Biorg. Med. Chem. 2018, 26, 4532–4536. [Google Scholar] [CrossRef]
- Hanuš, L.O.; Meyer, S.M.; Muñoz, E.; Taglialatela-Scafati, O.; Appendino, G. Phytocannabinoids: A unified critical inventory. Nat. Prod. Rep. 2016, 33, 1357–1392. [Google Scholar] [CrossRef]
- Nelson, K.M.; Bisson, J.; Singh, G.; Graham, J.G.; Chen, S.-N.; Friesen, J.B.; Dahlin, J.L.; Niemitz, M.; Walters, M.A.; Pauli, G.F. The Essential Medicinal Chemistry of Cannabidiol (CBD). J. Med. Chem. 2020, 63, 12137–12155. [Google Scholar] [CrossRef]
- Hanuš, L.O.; Tchilibon, S.; Ponde, D.E.; Breuer, A.; Fride, E.; Mechoulam, R. Enantiomeric cannabidiol derivatives: Synthesis and binding to cannabinoid receptors. Org. Biomol. Chem. 2005, 3, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Niyangoda, D.; Aung, M.L.; Qader, M.; Tesfaye, W.; Bushell, M.; Chiong, F.; Tsai, D.; Ahmad, D.; Samarawickrema, I.; Sinnollareddy, M.; et al. Cannabinoids as antibacterial agents: A systematic and critical review of in vitro efficacy against Streptococcus and Staphylococcus. Antibiotics 2024, 13, 1023. [Google Scholar] [CrossRef] [PubMed]
- Sahm, D.F.; Kissinger, J.; Gilmore, M.S.; Murray, P.R.; Mulder, R.; Solliday, J.; Clarke, B. In vitro susceptibility studies of vancomycin-resistant Enterococcus faecalis. Antimicrob. Agents Chemother. 1989, 33, 1588–1591. [Google Scholar] [CrossRef] [PubMed]
- Schleifer, K.H.; Kilpper-Bälz, R. Transfer of Streptococcus faecalis and Streptococcus faecium to the genus Enterococcus nom. rev. as Enterococcus faecalis comb. nov. and Enterococcus faecium comb. nov. Int. J. Syst. Evol. Microbiol. 1984, 34, 31–34. [Google Scholar] [CrossRef]
- Bugg, T.D.; Wright, G.D.; Dutka-Malen, S.; Arthur, M.; Courvalin, P.; Walsh, C.T. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: Biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry 1991, 30, 10408–10415. [Google Scholar] [CrossRef]
- Allegra, E.; Titball, R.W.; Carter, J.; Champion, O.L. Galleria mellonella larvae allow the discrimination of toxic and non-toxic chemicals. Chemosphere 2018, 198, 469–472. [Google Scholar] [CrossRef]
- Cortes, E.; Mora, J.; Márquez, E. Modelling the Anti-Methicillin-Resistant Staphylococcus Aureus (MRSA) Activity of Cannabinoids: A QSAR and Docking Study. Crystals 2020, 10, 692. [Google Scholar] [CrossRef]
- Chika, A.; Otalike, E.; Lawal, S.; Umar, M.; Usman, A.; Amali, A.; Yahaya, M.; Adamu, A. Repositioning of FDA-approved drugs for the treatment of methicillin-resistant Staphylococcus aureus infection: Structure-based virtual screening and in vitro assay. Natl. J. Physiol. Pharm. Pharmacol. 2023, 13, 813–819. [Google Scholar] [CrossRef]
- De Vita, S.; Lauro, G.; Ruggiero, D.; Terracciano, S.; Riccio, R.; Bifulco, G. Protein Preparation Automatic Protocol for High-Throughput Inverse Virtual Screening: Accelerating the Target Identification by Computational Methods. J. Chem. Inf. Model. 2019, 59, 4678–4690. [Google Scholar] [CrossRef]
- Schiebel, J.; Chang, A.; Lu, H.; Baxter, M.V.; Tonge, P.J.; Kisker, C. Staphylococcus aureus FabI: Inhibition, substrate recognition, and potential implications for in vivo essentiality. Structure 2012, 20, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshi, A.; Kim, E.E.; Hwang, K.Y. Structural insights into Staphylococcus aureus enoyl-ACP reductase (FabI), in complex with NADP and triclosan. Proteins Struct. Funct. Bioinform. 2010, 78, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Campobasso, N.; Patel, M.; Wilding, I.E.; Kallender, H.; Rosenberg, M.; Gwynn, M.N. Staphylococcus aureus 3-Hydroxy-3-methylglutaryl-CoA Synthase: Crystal structure and mechanism. J. Biol. Chem. 2004, 279, 44883–44888. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, P.K.; Dawson, A.; Hutchison, M.-T.; Cameron, S.; Hunter, W.N. Structure of Staphylococcus aureus adenylosuccinate lyase (PurB) and assessment of its potential as a target for structure-based inhibitor discovery. Acta Crystallogr. Sect. D. Biol. Crystallogr. 2010, 66, 881–888. [Google Scholar] [CrossRef]
- Zakataeva, N.P. Microbial 5′-nucleotidases: Their characteristics, roles in cellular metabolism, and possible practical applications. Appl. Microbiol. Biotechnol. 2021, 105, 7661–7681. [Google Scholar] [CrossRef]
- Blaszczyk, J.; Li, Y.; Gan, J.; Yan, H.; Ji, X. Structural Basis for the Aldolase and Epimerase Activities of Staphylococcus aureus Dihydroneopterin Aldolase. J. Mol. Biol. 2007, 368, 161–169. [Google Scholar] [CrossRef]
- Wang, S.; Reeve, S.M.; Holt, G.T.; Ojewole, A.A.; Frenkel, M.S.; Gainza, P.; Keshipeddy, S.; Fowler, V.G.; Wright, D.L.; Donald, B.R. Chiral evasion and stereospecific antifolate resistance in Staphylococcus aureus. PLoS Comp. Biol. 2022, 18, e1009855. [Google Scholar] [CrossRef]
- Pradhan, S.; Sinha, C. High throughput screening against pantothenate synthetase identifies amide inhibitors against Mycobacterium tuberculosis and Staphylococcus aureus. Silico Pharmacol. 2018, 6, 9. [Google Scholar] [CrossRef]
- Patel, K.M.; Teran, D.; Zheng, S.; Kandale, A.; Garcia, M.; Lv, Y.; Schembri, M.A.; McGeary, R.P.; Schenk, G.; Guddat, L.W. Crystal Structures of Staphylococcus aureus Ketol-Acid Reductoisomerase in Complex with Two Transition State Analogues that Have Biocidal Activity. Chemistry—A Eur. J. 2017, 23, 18289–18295. [Google Scholar] [CrossRef]
- Chen, C.; Joo Jeong, C.; Brown, G.; Stolnikova, E.; Halavaty Andrei, S.; Savchenko, A.; Anderson Wayne, F.; Yakunin Alexander, F. Structure-Based Mutational Studies of Substrate Inhibition of Betaine Aldehyde Dehydrogenase BetB from Staphylococcus aureus. Appl. Environ. Microbiol. 2014, 80, 3992–4002. [Google Scholar] [CrossRef]
- Wallace, B.D.; Edwards, J.S.; Wallen, J.R.; Moolman, W.J.A.; van der Westhuyzen, R.; Strauss, E.; Redinbo, M.R.; Claiborne, A. Turnover-Dependent Covalent Inactivation of Staphylococcus aureus Coenzyme A-Disulfide Reductase by Coenzyme A-Mimetics: Mechanistic and Structural Insights. Biochemistry 2012, 51, 7699–7711. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Chen, B.; Du, Z.; Zhao, B.; Li, J.; Li, Z.; Arunachalam, K.; Shi, T.; Wei, D.; Shi, C. Eugenol targeting CrtM inhibits the biosynthesis of staphyloxanthin in Staphylococcus aureus. Food Sci. Hum. Wellness 2024, 13, 1368–1377. [Google Scholar] [CrossRef]
- Shearer, H.L.; Loi, V.V.; Weiland, P.; Bange, G.; Altegoer, F.; Hampton, M.B.; Antelmann, H.; Dickerhof, N. MerA functions as a hypothiocyanous acid reductase and defense mechanism in Staphylococcus aureus. Mol. Microbiol. 2023, 119, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Skaar, E.P.; Gaspar, A.H.; Schneewind, O. IsdG and IsdI, Heme-degrading Enzymes in the Cytoplasm of Staphylococcus aureus. J. Biol. Chem. 2004, 279, 436–443. [Google Scholar] [CrossRef]
- Chetri, S. The culmination of multidrug-resistant efflux pumps vs. meager antibiotic arsenal era: Urgent need for an improved new generation of EPIs. Front. Microbiol. 2023, 14, 1149418. [Google Scholar] [CrossRef]
- Makino, Y.; Oe, C.; Iwama, K.; Suzuki, S.; Nishiyama, A.; Hasegawa, K.; Okuda, H.; Hirata, K.; Ueno, M.; Kawaji, K.; et al. Serine hydroxymethyltransferase as a potential target of antibacterial agents acting synergistically with one-carbon metabolism-related inhibitors. Commun. Biol. 2022, 5, 619. [Google Scholar] [CrossRef]
- Brötz-Oesterhelt, H.; Sass, P. Bacterial caseinolytic proteases as novel targets for antibacterial treatment. Int. J. Med. Microbiol. 2014, 304, 23–30. [Google Scholar] [CrossRef]
- Motiwala, T.; Mthethwa, Q.; Achilonu, I.; Khoza, T. ESKAPE Pathogens: Looking at Clp ATPases as Potential Drug Targets. Antibiotics 2022, 11, 1218. [Google Scholar] [CrossRef]
- Reissier, S.; Cattoir, V. Streptogramins for the treatment of infections caused by Gram-positive pathogens. Expert Rev. Anti Infect. Ther. 2021, 19, 587–599. [Google Scholar] [CrossRef]
- Sugantino, M.; Roderick, S.L. Crystal Structure of Vat(D): An Acetyltransferase That Inactivates Streptogramin Group A Antibiotics. Biochemistry 2002, 41, 2209–2216. [Google Scholar] [CrossRef]
- Cho, H.Y.; Nam, M.S.; Hong, H.J.; Song, W.S.; Yoon, S.-I. Structural and Biochemical Analysis of the Furan Aldehyde Reductase YugJ from Bacillus subtilis. Int. J. Mol. Sci. 2022, 23, 1882. [Google Scholar] [CrossRef] [PubMed]
- Pulingam, T.; Parumasivam, T.; Gazzali, A.M.; Sulaiman, A.M.; Chee, J.Y.; Lakshmanan, M.; Chin, C.F.; Sudesh, K. Antimicrobial resistance: Prevalence, economic burden, mechanisms of resistance and strategies to overcome. Eur. J. Pharm. Sci. 2022, 170, 106103. [Google Scholar] [CrossRef] [PubMed]
- Rana, P.; Ghouse, S.M.; Akunuri, R.; Madhavi, Y.V.; Chopra, S.; Nanduri, S. FabI (enoyl acyl carrier protein reductase)—A potential broad spectrum therapeutic target and its inhibitors. Eur. J. Med. Chem. 2020, 208, 112757. [Google Scholar] [CrossRef] [PubMed]
- Douglas, E.J.A.; Wulandari, S.W.; Lovell, S.D.; Laabei, M. Novel antimicrobial strategies to treat multi-drug resistant Staphylococcus aureus infections. Microb. Biotechnol. 2023, 16, 1456–1474. [Google Scholar] [CrossRef]
- Hayashi, H.; Saijo, E.; Hirata, K.; Murakami, S.; Okuda, H.; Kodama, E.N.; Hasegawa, K.; Murayama, K. SHIN-2 exerts potent activity against VanA-type vancomycin-resistant Enterococcus faecium in vitro by stabilizing the active site loop of serine hydroxymethyltransferase. Arch. Biochem. Biophys. 2024, 761, 110160. [Google Scholar] [CrossRef]
- Abu Lila, A.S.; Alharby, T.N.; Alanazi, J.; Alanazi, M.; Abdallah, M.H.; Rizvi, S.M.D.; Moin, A.; Khafagy, E.S.; Tabrez, S.; Al Balushi, A.A.; et al. Clinical resistant strains of Enterococci and their correlation to reduced susceptibility to biocides: Phenotypic and genotypic analysis of macrolides, lincosamides, and atreptogramins. Antibiotics 2023, 12, 461. [Google Scholar] [CrossRef]
- Gargiulo, E.; Moriello, A.S.; Benetti, E.; Pagni, L.; Arnoldi, L.; De Petrocellis, L.; Chianese, G.; Vitale, R.M.; Taglialatela-Scafati, O. Phytochemical characterization and TRPA1/TRPM8 modulation profile of the cannabigerol-rich Cannabis sativa L. Chemotype IV. J. Nat. Prod. 2024, 87, 722–732. [Google Scholar] [CrossRef]
- D’Agostino, I.; Ardino, C.; Poli, G.; Sannio, F.; Lucidi, M.; Poggialini, F.; Visaggio, D.; Rango, E.; Filippi, S.; Petricci, E.; et al. Antibacterial alkylguanidino ureas: Molecular simplification approach, searching for membrane-based MoA. Eur. J. Med. Chem. 2022, 231, 114158. [Google Scholar] [CrossRef]
- Schrödinger Release 2024-1: Maestro; Schrödinger LLC: New York, NY, USA, 2024.
- Schrödinger Release 2024-1: LigPrep; Schrödinger LLC: New York, NY, USA, 2024.
- Schrödinger Release 2024-1: Protein Preparation Wizard; Schrödinger LLC: New York, NY, USA, 2024.
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Schrödinger Release 2024-1: SiteMap; Schrödinger LLC: New York, NY, USA, 2024.
- Schrödinger Release 2024-1: Glide; Schrödinger LLC: New York, NY, USA, 2024.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
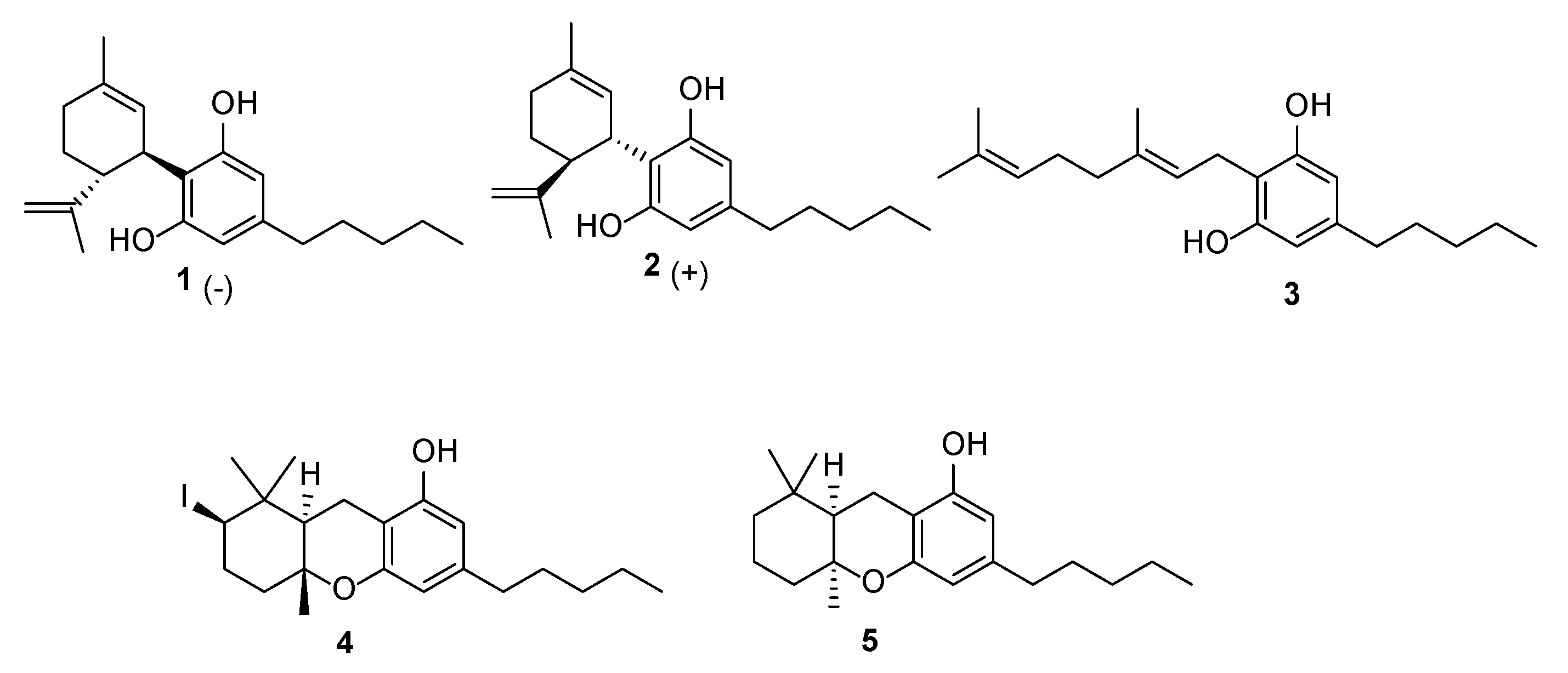

{kind=link}
{kind=link}
{kind=link}
| Strain | Gram | Country | Year | Isolation Source | Resistance | References | |
|---|---|---|---|---|---|---|---|
| Bacillus subtilis subsp. spizizenii | ATCC 6633 | + | ns | 1976 | desert soil | NIS | American Type Culture Collection |
| Escherichia coli | MG1655 | - | ns | 1895 | laboratory strain | ns | American Type Culture Collection |
| Enterococcus faecalis | ATCC 29212 | + | ns | ns | urine | ns | American Type Culture Collection |
| Enterococcus faecalis | ATCC 700802 | + | USA | 1987 | blood | VAN (vanB), GEN | [24] |
| Enterococcus faecium | ATCC 19434 T | + | ns | ns | ns | ns | [25] |
| Enterococcus faecium | BM4147 | + | ns | ns | ns | VAN (vanA) | [26] |
| Staphylococcus aureus | ATCC 25923 | + | USA | 1945 | ns | MSSA | American Type Culture Collection |
| Staphylococcus aureus | ATCC 43300 | + | USA | ns | ns | MRSA | American Type Culture Collection |
| MIC (µg/mL) | MBC (µg/mL) | |||||||
|---|---|---|---|---|---|---|---|---|
| Cpd | S. aureus ATCC 25923 | S. aureus ATCC 43300 | E. faecalis ATCC 29212 | E. faecalis ATCC 700802 | E. faecium ATCC 19434 | E. faecium BM4147 | B. spizizenii DSM 347 | S. aureus ATCC 25923 |
| 1 | 2 | 2 | 4 | 4 | 1 | 2 | 2 | 8 |
| 2 | 2 | 8 | 8 | 8 | 2 | 1 | 4 | 8 |
| 3 | 2 | 1 | 2 | 2 | 1 | 1 | 1 | 4 |
| 4 | 8 | >64 | 4 | 8 | 2 | 1 | 4 | >64 |
| 5 | 2 | 4 | 4 | 4 | 2 | 1 | 2 | 8 |
| Compound | Target Proteins |
|---|---|
| 1 | I3U4H4, P50870, Q3XX76, Q3Y316, Q9WVY4 |
| 2 | I3U4H4, P50870, Q3XX76, Q3Y316 |
| 3 | A0A1S8KJG1, I3U4H4, P50870, Q3XX76 |
| 4 | P50870 |
| 5 | A0A1S8KJG1, I3U4H4, P50870 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirignano, C.; De Vita, S.; Gargiulo, E.; Lucidi, M.; Visaggio, D.; Chini, M.G.; Lauro, G.; Chianese, G.; Visca, P.; Bifulco, G.; et al. Exploring the Potential of Phytocannabinoids Against Multidrug-Resistant Bacteria. Plants 2025, 14, 1901. https://doi.org/10.3390/plants14131901
Sirignano C, De Vita S, Gargiulo E, Lucidi M, Visaggio D, Chini MG, Lauro G, Chianese G, Visca P, Bifulco G, et al. Exploring the Potential of Phytocannabinoids Against Multidrug-Resistant Bacteria. Plants. 2025; 14(13):1901. https://doi.org/10.3390/plants14131901
Chicago/Turabian StyleSirignano, Carmina, Simona De Vita, Ernesto Gargiulo, Massimiliano Lucidi, Daniela Visaggio, Maria Giovanna Chini, Gianluigi Lauro, Giuseppina Chianese, Paolo Visca, Giuseppe Bifulco, and et al. 2025. "Exploring the Potential of Phytocannabinoids Against Multidrug-Resistant Bacteria" Plants 14, no. 13: 1901. https://doi.org/10.3390/plants14131901
APA StyleSirignano, C., De Vita, S., Gargiulo, E., Lucidi, M., Visaggio, D., Chini, M. G., Lauro, G., Chianese, G., Visca, P., Bifulco, G., & Taglialatela-Scafati, O. (2025). Exploring the Potential of Phytocannabinoids Against Multidrug-Resistant Bacteria. Plants, 14(13), 1901. https://doi.org/10.3390/plants14131901