1. Introduction
Pseudomonas savastanoi pv.
savastanoi is the causal agent of olive knot disease (OKD), characterized by the formation of tumorous growths, or knots, primarily on the branches and twigs of olive trees (
Olea europaea) [
1,
2], and occasionally on leaves and fruits [
2,
3,
4,
5]. OKD is a chronic disease, as symptoms persist and recur for many years in infected plants.
P. savastanoi pv.
savastanoi enters the host plant through wounds resulting from various causes such as harvesting, pruning, hail, frost, and leaf scars. Knot development is driven by bacterial phytohormones, such as 3-indoleacetic acid and cytokinins, which promote uncontrolled cell growth around the infection site [
6,
7,
8]. The knots contribute to the decline and death of branches, resulting in significant yield losses. Tree vigour, growth and the size and quality of the fruits can be moderately or severely reduced [
1,
9]. Crop losses caused by olive knot are not clearly assessed, and greatly depend on the geographical location and olive cultivar [
4].
P. savastanoi pv.
savastanoi can persist as an epiphyte on the surface of aerial plant parts and within young knots [
1,
10]. This persistence represents a critical inoculum source for new infections, allowing for rapid disease transmission across the entire orchard [
1]. The bacterium can spread via rain, wind, insects, and human activities, such as olive grove management practices.
The dynamics of bacterial populations are influenced by seasonal factors and leaf age, with the most significant damage occurring under conditions that favor
P. savastanoi pv.
savastanoi epiphytic growth and its entry into olive bark, particularly during spring and autumn compared to winter and summer [
10,
11,
12]. High rainfall and moderate temperatures (10–20 °C) are optimal for
P. savastanoi pv.
savastanoi epiphytic growth and penetration, resulting in significant harm to the host plant [
12,
13,
14]. The spread of the disease through asymptomatic propagation materials is well-documented, as the epiphytic and endophytic lifestyle of
P. savastanoi pv.
savastanoi makes plants for planting a key pathway for its long-distance dissemination [
15,
16,
17]. Consequently, the pathogen is usually introduced into new areas through asymptomatic infected plant material.
Controlling OKD is challenging, and current strategies focus on preventive measures such as reducing both endophytic and epiphytic bacterium populations through sanitary and cultural practices [
1,
3,
4,
18]. These strategies include pruning symptomatic branches, disinfecting the resulting wound and applying foliar treatments with copper-based compounds. Preventive measures also include the use of certified pathogen-tested plants and rootstocks for establishing new olive groves [
19]. It is worth noting that the European phytosanitary certification program for the production of certified olive trees includes verification of the absence of
P. savastanoi pv.
savastanoi [
20]. Thus, reliable diagnostic tests for detecting
P. savastanoi pv.
savastanoi in asymptomatic plant material are essential. Conventional
P. savastanoi pv.
savastanoi diagnosis relies on visual inspection of typical symptoms (e.g., knots) and direct bacterial isolation followed by pathogenicity and biochemical or serological tests [
21,
22,
23,
24]. However, these methods are time-consuming and lack sensitivity and specificity, especially for asymptomatic plants, where bacterial isolation is often unreliable [
25,
26,
27,
28,
29]. To overcome these limitations, several assays for
P. savastanoi pv.
savastanoi detection have been developed, including PCR [
26], nested PCR [
29], enriched PCR [
26], qPCR [
27], and High-Resolution Melting Analysis (HRMA) [
30]. These tools have demonstrated sensitivity and specificity, as well as the ability to discriminate among different pathovars (i.e.,
savastanoi,
nerii,
fraxini) or to quantify the pathogen. Despite this, a low bacterial load and/or the presence of inhibitors in plant matrices may lead to false negatives even with molecular methods. Under these conditions, two steps could be improved: (i) plant material processing to increase the detectable bacterial load and (ii) molecular tests less affected by inhibitors.
Bertolini et al. (2003) [
28] compared two methods for processing plant material: external washing versus washing followed by an enrichment step. Their findings showed that the enrichment step enhanced the detection efficiency of
P. savastanoi pv.
savastanoi, both as an epiphyte and an endophyte. In this study, the initial steps for processing plant material were optimized to improve bacterial DNA detection by increasing sensitivity and minimizing PCR inhibition. To achieve this, the protocol described by Bertolini et al. 2003 [
28] was partially modified by adding a concentration step after washing and by reducing the enrichment time from 72 h to 24 or 48 h. DNA was then extracted using four different approaches: (i) external washing step (EW); (ii) pellet of concentrated washing (PELLET); (iii) 24 h enrichment (24hE); and (iv) 48 h enrichment (48hE). These preparations were tested using both qPCR [
27] and droplet digital PCR (ddPCR). The ddPCR has shown promise for efficiently detecting pathogens such as
Ralstonia solanacearum in potatoes [
31] and
Xylella fastidiosa spp. in several plant hosts [
32], showing sensitivity comparable to qPCR. Other studies showed increased detection sensitivity of ddPCR compared to qPCR for
Xanthomonas citri subsp.
citri [
33,
34], pepper mild mottle virus in plants, soil, and water [
35], and
Phytophthora nicotianae in environmental samples [
36]. Recently, a ddPCR protocol for
X. fastidiosa detection was implemented and showed greater analytical sensitivity than qPCR for
O. europea, C. sinensis, and
N. oleander [
37]. The ddPCR allows the detection and quantification of pathogens, such as
Agrobacterium vitis in grapevines, for which previous methods lacked sensitivity [
38]. Unlike qPCR, ddPCR provides absolute target quantification without the need for a standard curve, facilitating data comparison and experimental design when standards are unavailable [
38]. Additionally, ddPCR is less affected by PCR inhibitors than qPCR [
38,
39]. Given the challenges of detecting
P. savastanoi pv.
savastanoi in asymptomatic samples, using ddPCR as a highly sensitive and specific diagnostic method, this study aimed to develop a protocol for preparing samples and adapting the qPCR method by Tegli et al. 2010 [
27] to ddPCR. To achieve this, ddPCR was first developed using spiked samples and then tested on naturally infected asymptomatic samples collected from olive trees in different Italian regions. The developed procedure showed high sensitivity, representing a valid diagnostic tool for testing the phytosanitary status of olive plant material.
3. Discussion
Olive knot disease is known for causing severe damage to olive trees, significantly affecting both the yield and quality of olive oil, with substantial economic consequences for the Mediterranean olive oil industry [
2].
The pathogen’s ability to persist, even in a low concentration, in asymptomatic olive material represents a critical challenge for the management of OKD and for the detection of the causal agent
P. savastanoi pv.
savastanoi. This bacterium thrives in the Mediterranean climate, where conditions such as high humidity and moderate temperatures promote its growth [
12,
13,
14].
Conventional control strategies for OKD primarily focus on reducing bacterial populations through physical and chemical means. Pruning infected branches helps remove visible sources of infection [
3], while traditional copper-based treatments or innovative approaches, such as copper-nanoparticles, thyme essential oil nanoparticles [
40,
41], or essential oils [
42] aim to reduce the bacterial load. In addition to implementing control measures, early and accurate detection of
P. savastanoi pv.
savastanoi is essential to prevent the spread and introduction in new areas of
P. savastanoi pv.
savastanoi. Therefore, the application of efficient and reliable diagnostic methods becomes crucial. The current diagnostic tools, including visual inspection, isolation and qPCR, have limitations. Visual inspection and bacterial isolation can be unreliable, especially in asymptomatic plants where disease symptoms are not yet evident and bacterial load is generally low. Although qPCR is a highly sensitive molecular test, it can be not efficient to detect low bacterial loads in asymptomatic samples and may be affected by inhibitors present in plant matrices [
43,
44,
45]. This highlights the need for more reliable diagnostic tools that can detect the pathogen before it leads to noticeable symptoms and, consequently, damage.
In this context ddPCR shows increased sensitivity and specificity by partitioning the sample into thousands of droplets, allowing for precise quantification of target DNA without relying on a standard curve [
38,
45]. This makes ddPCR particularly suited for detecting pathogens at low concentrations, which is essential for identifying latent infections in asymptomatic plants [
36,
46].
This study focused on the optimization of the procedure for the detection of P. savastanoi pv. savastanoi from an asymptomatic olive plant, by improving sample preparation and by developing a sensitive ddPCR to enhance diagnostic reliability.
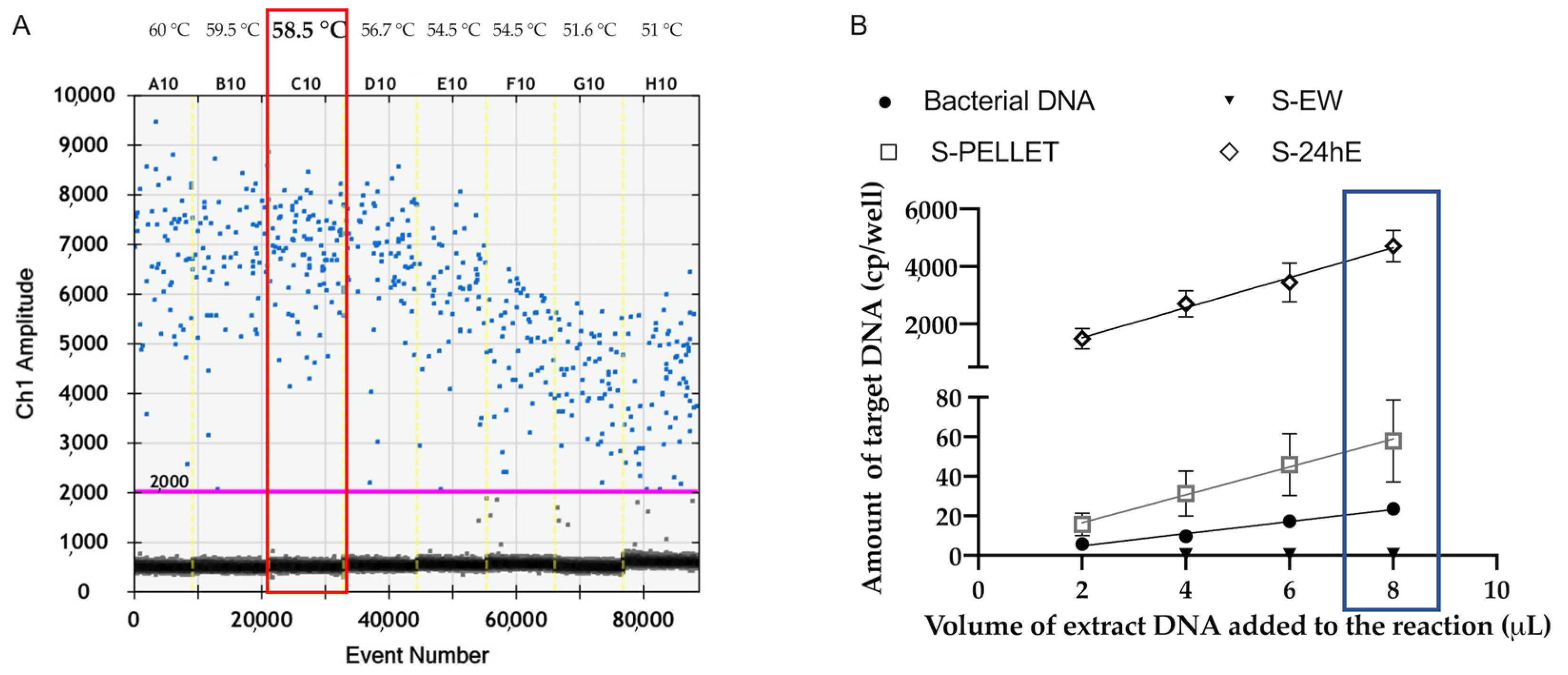
Among the four evaluated preparations on spiked samples, the S-PELLET and S-24hE proved to be the most effective in terms of analytical sensitivity. Furthermore, the S-PELLET, which involves concentrating the bacterial load of the washing solution, showed higher sensitivity achieving a high limit of detection of 10 CFU/mL for both qPCR PCR and ddPCR. Conversely, the S-EW exhibited lower sensitivity, likely due to the low bacterial load in the washing solution fraction. The S-48hE preparation, while effective for P. savastanoi pv. savastanoi detection by qPCR, showed ddPCR saturation, even at the lower concentration of 10 CFU/mL, making it less suitable for ddPCR.
Regarding the two selected sample preparation methods (F-PELLET and F-24hE) used for the analysis of field samples, ddPCR demonstrated higher efficiency than qPCR in detecting P. savastanoi pv. savastanoi in 100 asymptomatic samples collected from both symptomatic and asymptomatic olive trees. These results highlight the improved analytical sensitivity of ddPCR, especially under challenging conditions where bacterial concentrations may be low, unevenly distributed, or where cells may be in a viable but non-culturable (VBNC) state—common scenarios in asymptomatic plant tissues.
Unlike spiked samples, field samples present greater variability. Such variability can affect both the presence and composition of PCR inhibitors. In this context, ddPCR offers a clear advantage over qPCR, as its droplet-based partitioning reduces the impact of inhibitors and enhances the detection of low bacterial DNA concentrations, thereby increasing overall accuracy. The higher efficiency of ddPCR in detecting the F-PELLET samples can be also attributed to its ability to detect DNA from both viable and non-viable bacterial cells, with respect to F-24hE preparation that enriched only viable cells. The concentration step increases the likelihood of capturing bacterial cells, even when the bacterial load is low. This makes it particularly useful for detecting
P. savastanoi pv.
savastanoi in asymptomatic samples, where bacteria may be present in low numbers or in a latent state. Conversely, the F-24hE, which requires viable bacterial cells to grow in an enrichment medium, was less effective. The F-24hE may be less effective than F-PELLET also due to the presence of
P. savastanoi pv.
savastanoi in a VBNC state.
P. savastanoi pv.
savastanoi can switch from a viable to a VBNC state, i.e., in plants exposed to heavy metal copper ions [
18]. However, the F-24hE procedure still provided valuable insights when combined with ddPCR.
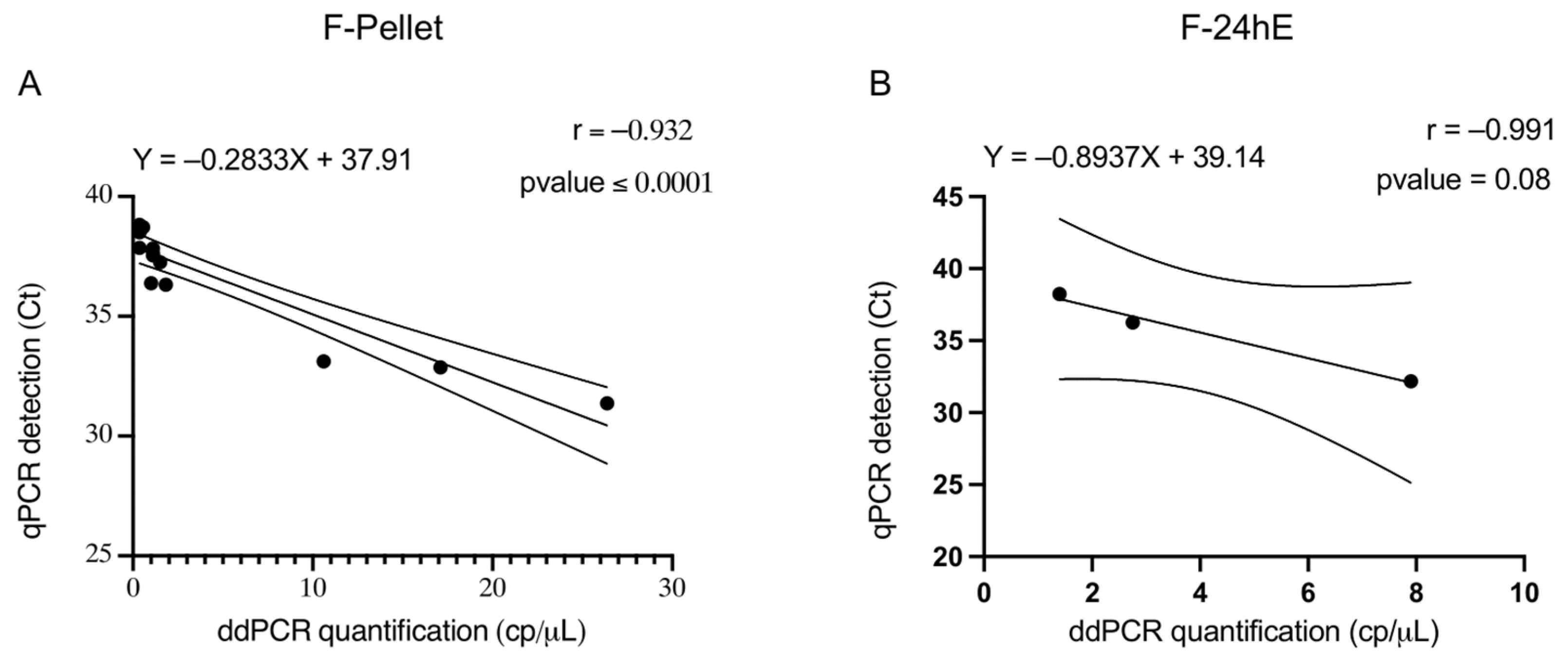
Pearson’s correlation coefficient for PELLET samples was statistically significant, indicating a strong correlation between the results obtained with the two molecular methods. This high correlation supports the reliability of ddPCR as an alternative to qPCR, with the added benefit of increased efficiency and sensitivity.
Given the seasonality of P. savastanoi pv. savastanoi’s activity and epiphytic growth we investigated if monitoring activity may be affected by the season of sampling. Our results highlighted a higher efficiency of ddPCR compared to qPCR, particularly during April/May for both sample preparation methods (F-PELLET: 40% vs. 12%; F-24hE: 31% vs. 4%). Nevertheless, these differences were not statistically significant. During the October/November sampling period, ddPCR also showed higher performance than qPCR, although the differences were less pronounced compared to the April/May period (F-PELLET: 26% vs. 20%; F-24hE: 13% vs. 2%). Similarly, these differences were not statistically significant.
In conclusion, the findings of the spiked and the field samples confirm that ddPCR is a highly sensitive and reliable tool for detecting P. savastanoi pv. savastanoi in olive trees, particularly in asymptomatic plant material. The PELLET preparation, in combination with ddPCR, offers the most effective procedure. On the other hand, the 24hE can be useful when the detection of only viable cells is advisable. Together, these results highlight the potential of ddPCR to improve early detection and management of OKD, ultimately contributing to the prevention and limiting the spread of this harmful pathogen through asymptomatic plant propagation material.


 ,
, 

{kind=link}
{kind=link}
{kind=link}