
Evolutionary Dynamics of Plant TRM6/TRM61 Complexes


Abstract
:1. Introduction
2. Results
2.1. Identification of TRM6 and TRM61 Families in Plants
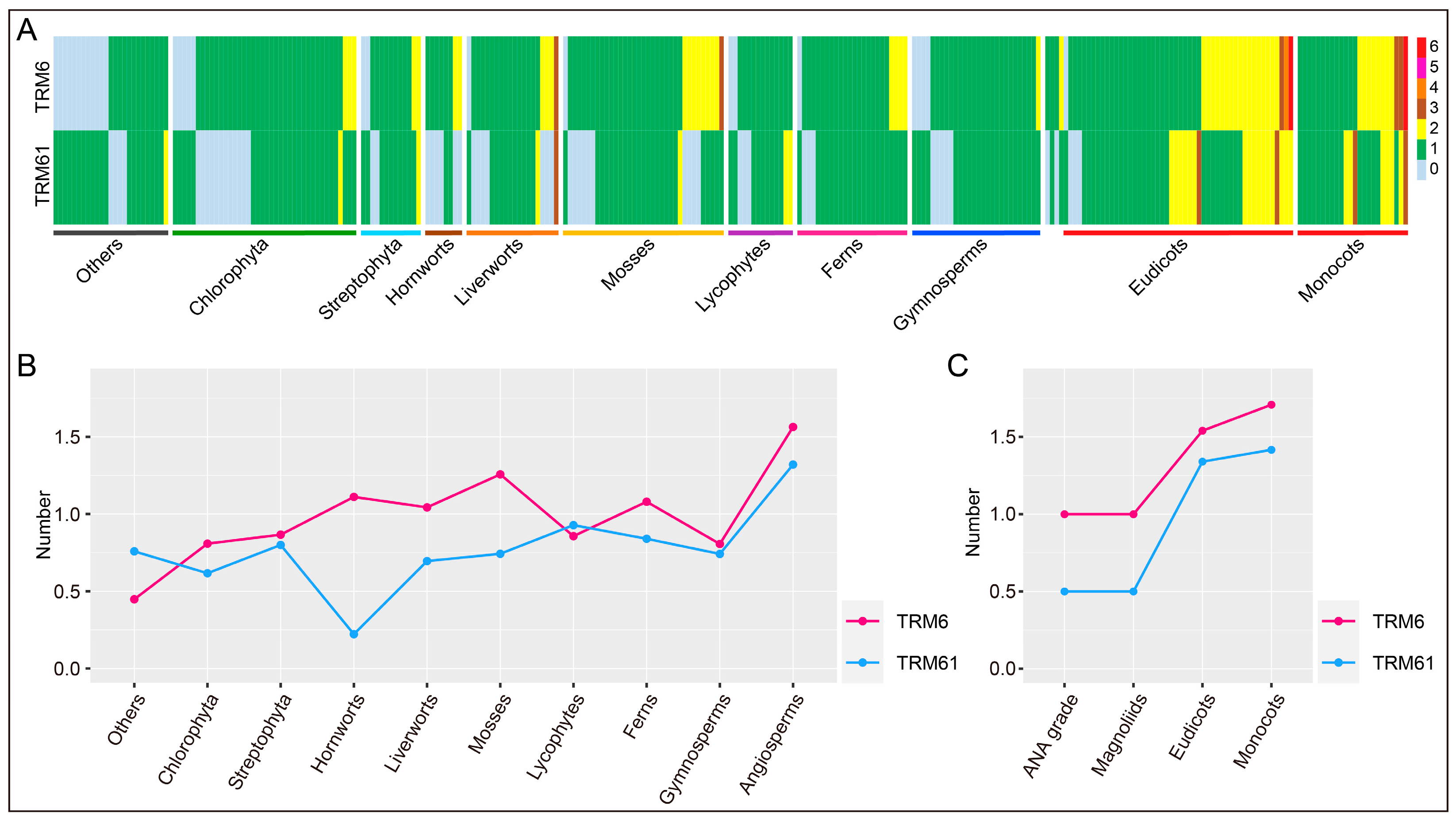
2.2. The Evolution of TRM6 and TRM61 Families in Plants
2.3. Conserved TRM6 and TRM61 Homologs Show Similar Expression Profiles in Plants
2.4. The Evolutionary Dynamics of TRM6 and TRM61 Homologs in the Wheat Lineage
2.5. TRM6 and TRM61 Homologs Undergo Divergent Selection Pressures During Wheat Evolution and Breeding
2.6. Divergent Expression Profiles of TRM6/TRM61 Homologs in Wheat
3. Discussion
3.1. Subunit Plasticity and Structural Innovation in Plant m1A Methyltransferases
3.2. Evolutionary Conservation and Functional Divergence of TRM6 and TRM61 Homologs in Plants
3.3. The Evolutionary Fate of Duplicated TRM6 and TRM61 Genes in Wheat
4. Materials and Methods
4.1. Species Classification and Data Collection
4.2. TRM6 and TRM61 Identification
4.3. Phylogenetic Analysis of the TRM6 and TRM61 Families
4.4. Expression Pattern and Conserved Motif Analysis of TRM6 and TRM61 Homologs in Phylogenetically Representative Plants
4.5. The Evolutionary Dynamics Analysis of TRM6 and TRM61 Families in the Wheat System
4.6. The Expression Profile Analysis and Interaction Prediction of TRM6 and TRM61 Homologs in Wheat
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Prall, W.; Bhatia, G.; Gregory, B.D. The diversity and functions of plant RNA modifications: What we know and where we go from here. Annu. Rev. Plant Biol. 2023, 74, 53–85. [Google Scholar] [CrossRef]
- McCown, P.J.; Ruszkowska, A.; Kunkler, C.N.; Breger, K.; Hulewicz, J.P.; Wang, M.C.; Springer, N.A.; Brown, J.A. Naturally occurring modified ribonucleosides. Wiley Interdiscip. Rev. RNA 2020, 11, e1595. [Google Scholar] [CrossRef] [PubMed]
- Cappannini, A.; Ray, A.; Purta, E.; Mukherjee, S.; Boccaletto, P.; Moafinejad, S.N.; Lechner, A.; Barchet, C.; Klaholz, B.P.; Stefaniak, F.; et al. MODOMICS: A database of RNA modifications and related information. 2023 update. Nucleic Acids Res. 2024, 52, D239–D244. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Feng, T.; Yuan, B.F. Advances in mapping analysis of ribonucleic acid modifications through sequencing. Chin. J. Chromatogr. 2024, 42, 632–645. [Google Scholar] [CrossRef]
- Xuan, J.J.; Sun, W.J.; Lin, P.H.; Zhou, K.R.; Liu, S.; Zheng, L.L.; Qu, L.H.; Yang, J.H. RMBase v2.0: Deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018, 46, D327–D334. [Google Scholar] [CrossRef]
- Grosjean, H. RNA modification: The Golden Period 1995-2015. RNA 2015, 21, 625–626. [Google Scholar] [CrossRef]
- Kramer, M.C.; Anderson, S.J.; Gregory, B.D. The nucleotides they are a-changin’: Function of RNA binding proteins in post-transcriptional messenger RNA editing and modification in Arabidopsis. Curr. Opin. Plant Biol. 2018, 45, 88–95. [Google Scholar] [CrossRef]
- Yu, X.; Sharma, B.; Gregory, B.D. The impact of epitranscriptomic marks on post-transcriptional regulation in plants. Brief. Funct. Genom. 2021, 20, 113–124. [Google Scholar] [CrossRef]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef]
- Shen, L.; Liang, Z.; Wong, C.E.; Yu, H. Messenger RNA modifications in plants. Trends Plant Sci. 2019, 24, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.J.; Kramer, M.C.; Gosai, S.J.; Yu, X.; Vandivier, L.E.; Nelson, A.D.L.; Anderson, Z.D.; Beilstein, M.A.; Fray, R.G.; Lyons, E.; et al. N6-methyladenosine inhibits local ribonucleolytic cleavage to stabilize mRNAs in Arabidopsis. Cell Rep. 2018, 25, 1146–1157.e3. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Vicente, A.M.; Manavski, N.; Rohn, P.T.; Schmid, L.M.; Garcia-Molina, A.; Leister, D.; Seydel, C.; Bellin, L.; Mohlmann, T.; Ammann, G.; et al. The plant cytosolic m6A RNA methylome stabilizes photosynthesis in the cold. Plant Commun. 2023, 4, 100634. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Lv, R.; Ma, H.; Shen, H.; He, C.; Wang, J.; Jiao, F.; Liu, H.; Yang, P.; Tan, L.; et al. Zc3h13 regulates nuclear RNA m6A methylation and mouse embryonic stem cell self-renewal. Mol. Cell 2018, 69, 1028–1038.e1026. [Google Scholar] [CrossRef]
- Kontur, C.; Jeong, M.; Cifuentes, D.; Giraldez, A.J. Ythdf m6A readers function redundantly during zebrafish development. Cell Rep. 2020, 33, 108598. [Google Scholar] [CrossRef]
- Dunn, D.B. The occurrence of 1-methyladenine in ribonucleic acid. Biochim. Biophys. Acta 1961, 46, 198–200. [Google Scholar] [CrossRef]
- RajBhandary, U.L.; Stuart, A.; Faulkner, R.D.; Chang, S.H.; Khorana, H.G. Nucleotide sequence studies on yeast phenylalanine sRNA. Cold Spring Harb. Symp. Quant. Biol. 1966, 31, 425–434. [Google Scholar] [CrossRef]
- Sharma, S.; Watzinger, P.; Kotter, P.; Entian, K.D. Identification of a novel methyltransferase, Bmt2, responsible for the N-1-methyl-adenosine base modification of 25S rRNA in Saccharomyces cerevisiae. Nucleic Acids Res. 2013, 41, 5428–5443. [Google Scholar] [CrossRef]
- Li, X.; Xiong, X.; Wang, K.; Wang, L.; Shu, X.; Ma, S.; Yi, C. Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nat. Chem. Biol. 2016, 12, 311–316. [Google Scholar] [CrossRef]
- Zhou, H.; Rauch, S.; Dai, Q.; Cui, X.; Zhang, Z.; Nachtergaele, S.; Sepich, C.; He, C.; Dickinson, B.C. Evolution of a reverse transcriptase to map N1-methyladenosine in human messenger RNA. Nat. Methods 2019, 16, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Nachtergaele, S.; Moshitch-Moshkovitz, S.; Peer, E.; Kol, N.; Ben-Haim, M.S.; Dai, Q.; Di Segni, A.; Salmon-Divon, M.; Clark, W.C.; et al. The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature 2016, 530, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Cenik, C.; Chua, H.N.; Singh, G.; Akef, A.; Snyder, M.P.; Palazzo, A.F.; Moore, M.J.; Roth, F.P. A common class of transcripts with 5′-intron depletion, distinct early coding sequence features, and N1-methyladenosine modification. RNA 2017, 23, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Meng, J.; Liu, J.; Ding, B.; Tan, T.; Wei, Q.; Yu, Y. The N1-methyladenosine methylome of petunia mRNA. Plant Physiol. 2020, 183, 1710–1724. [Google Scholar] [CrossRef]
- Aslam, M.; Huang, X.Y.; Yan, M.K.; She, Z.Y.; Lu, X.Y.; Fakher, B.; Chen, Y.Z.; Li, G.; Qin, Y. TRM61 is essential for Arabidopsis embryo and endosperm development. Plant Reprod. 2022, 35, 31–46. [Google Scholar] [CrossRef]
- Tang, J.; Jia, P.F.; Xin, P.Y.; Chu, J.F.; Shi, D.Q.; Yang, W.C. The Arabidopsis TRM61/TRM6 complex is a bona fide tRNA N1-methyladenosine methyltransferase. J. Exp. Bot. 2020, 71, 3024–3036. [Google Scholar] [CrossRef]
- Ma, L.L.; Zuo, J.H.; Bai, C.M.; Fu, A.Z.; Wang, Q.; Zhou, Z.J.; Deng, Z.P.; Tan, J.J.; Bouzayen, M.; Zheng, Y.Y. The dynamic N1-methyladenosine RNA methylation provides insights into the tomato fruit ripening. Plant J. 2024, 120, 2014–2030. [Google Scholar] [CrossRef]
- Li, C.; Li, Y.; Guo, J.; Wang, Y.; Shi, X.; Zhang, Y.; Liang, N.; Ma, H.; Yuan, J.; Xu, J.; et al. Abundant mRNA m1A modification in dinoflagellates: A new layer of gene regulation. EMBO Rep. 2024, 25, 4655–4673. [Google Scholar] [CrossRef]
- Shao, Y.; Wong, C.E.; Shen, L.; Yu, H. N6-methyladenosine modification underlies messenger RNA metabolism and plant development. Curr. Opin. Plant Biol. 2021, 63, 102047. [Google Scholar] [CrossRef]
- Tang, J.; Chen, S.; Jia, G. Detection, regulation, and functions of RNA N6-methyladenosine modification in plants. Plant Commun. 2023, 4, 100546. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.W.; Kleiner, R.E. YTHDF2 recognition of N1-methyladenosine (m1A)-modified RNA is associated with transcript destabilization. ACS Chem. Biol. 2020, 15, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.X.; Wang, T.L.; Gonzalez, G.; Wang, Y.S. Identification of YTH domain-containing proteins as the readers for N1-methyladenosine in RNA. Anal. Chem. 2018, 90, 6380–6384. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Phan, L.; Cuesta, R.; Carlson, B.A.; Pak, M.; Asano, K.; Björk, G.R.; Tamame, M.; Hinnebusch, A.G. The essential Gcd10p-Gcd14p nuclear complex is required for 1-methyladenosine modification and maturation of initiator methionyl-tRNA. Gene Dev. 1998, 12, 3650–3662. [Google Scholar] [CrossRef]
- Anderson, J.; Phan, L.; Hinnebusch, A.G. The Gcd10p/Gcd14p complex is the essential two-subunit tRNA(1-methyladenosine) methyltransferase of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2000, 97, 5173–5178. [Google Scholar] [CrossRef]
- Wang, M.; Zhu, Y.; Wang, C.; Fan, X.; Jiang, X.; Ebrahimi, M.; Qiao, Z.; Niu, L.; Teng, M.; Li, X. Crystal structure of the two-subunit tRNA m1A58 methyltransferase TRM6-TRM61 from Saccharomyces cerevisiae. Sci. Rep. 2016, 6, 32562. [Google Scholar] [CrossRef]
- He, R.; Lv, Z.; Li, Y.; Ren, S.; Cao, J.; Zhu, J.; Zhang, X.; Wu, H.; Wan, L.; Tang, J.; et al. tRNA-m1A methylation controls the infection of Magnaporthe oryzae by supporting ergosterol biosynthesis. Dev. Cell 2024, 59, 2931–2946.e2937. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, L.; Mu, L.; Wang, Y.; Zhao, M.; Wang, H.; Li, X.; Zhao, L.; Lin, C.; Zhang, H.; et al. Evolution and post-transcriptional regulation insights of m6A writers, erasers, and readers in plant epitranscriptome. Plant J. 2024, 120, 505–525. [Google Scholar] [CrossRef]
- Marcussen, T.; Sandve, S.R.; Heier, L.; Spannagl, M.; Pfeifer, M.; Jakobsen, K.S.; Wulff, B.B.H.; Steuernagel, B.; Mayer, K.F.X.; Olsen, O.A.; et al. Ancient hybridizations among the ancestral genomes of bread wheat. Science 2014, 345, 1250092. [Google Scholar] [CrossRef]
- Wang, J.; Luo, M.C.; Chen, Z.; You, F.M.; Wei, Y.; Zheng, Y.; Dvorak, J. Aegilops tauschii single nucleotide polymorphisms shed light on the origins of wheat D-genome genetic diversity and pinpoint the geographic origin of hexaploid wheat. New Phytol. 2013, 198, 925–937. [Google Scholar] [CrossRef]
- Zhao, X.; Guo, Y.; Kang, L.; Yin, C.; Bi, A.; Xu, D.; Zhang, Z.; Zhang, J.; Yang, X.; Xu, J.; et al. Population genomics unravels the Holocene history of bread wheat and its relatives. Nat. Plants 2023, 9, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Roovers, M.; Wouters, J.; Bujnicki, J.M.; Tricot, C.; Stalon, V.; Grosjean, H.; Droogmans, L. A primordial RNA modification enzyme: The case of tRNA (m1A) methyltransferase. Nucleic Acids Res. 2004, 32, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Droogmans, L.; Roovers, M.; Bujnicki, J.M.; Tricot, C.; Hartsch, T.; Stalon, V.; Grosjean, H. Cloning and characterization of tRNA (m1A58) methyltransferase (TrmI) from Thermus thermophilus HB27, a protein required for cell growth at extreme temperatures. Nucleic Acids Res. 2003, 31, 2148–2156. [Google Scholar] [CrossRef] [PubMed]
- Ozanick, S.; Krecic, A.; Andersland, J.; Anderson, J.T. The bipartite structure of the tRNA m1A58 methyltransferase from S. cerevisiae is conserved in humans. RNA 2005, 11, 1281–1290. [Google Scholar] [CrossRef]
- Bujnicki, J.M. In silico analysis of the tRNA: m1A58 methyltransferase family: Homology-based fold prediction and identification of new members from Eubacteria and Archaea. FEBS Lett. 2001, 507, 123–127. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef]
- Vanneste, K.; Baele, G.; Maere, S.; Van de Peer, Y. Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous-Paleogene boundary. Genome Res. 2014, 24, 1334–1347. [Google Scholar] [CrossRef]
- Edger, P.P.; Pires, J.C. Gene and genome duplications: The impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res. 2009, 17, 699–717. [Google Scholar] [CrossRef]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Murat, F.; Armero, A.; Pont, C.; Klopp, C.; Salse, J. Reconstructing the genome of the most recent common ancestor of flowering plants. Nat. Genet. 2017, 49, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Freeling, M.; Scanlon, M.J.; Fowler, J.E. Fractionation and subfunctionalization following genome duplications: Mechanisms that drive gene content and their consequences. Curr. Opin. Genet. Dev. 2015, 35, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Conant, G.C.; Birchler, J.A.; Pires, J.C. Dosage, duplication, and diploidization: Clarifying the interplay of multiple models for duplicate gene evolution over time. Curr. Opin. Plant Biol. 2014, 19, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Birchler, J.A.; Veitia, R.A. Gene balance hypothesis: Connecting issues of dosage sensitivity across biological disciplines. Proc. Natl. Acad. Sci. USA 2012, 109, 14746–14753. [Google Scholar] [CrossRef]
- Li, Z.; Defoort, J.; Tasdighian, S.; Maere, S.; Van de Peer, Y.; De Smet, R. Gene duplicability of core genes is highly consistent across all angiosperms. Plant Cell 2016, 28, 326–344. [Google Scholar] [CrossRef]
- Qian, W.F.; Liao, B.Y.; Chang, A.Y.F.; Zhang, J.Z. Maintenance of duplicate genes and their functional redundancy by reduced expression. Trends Genet. 2010, 26, 425–430. [Google Scholar] [CrossRef]
- Yates, A.D.; Allen, J.; Amode, R.M.; Azov, A.G.; Barba, M.; Becerra, A.; Bhai, J.; Campbell, L.I.; Carbajo Martinez, M.; Chakiachvili, M.; et al. Ensembl Genomes 2022: An expanding genome resource for non-vertebrates. Nucleic Acids Res. 2022, 50, D996–D1003. [Google Scholar] [CrossRef]
- One Thousand Plant Transcriptomes, I. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 2019, 574, 679–685. [Google Scholar] [CrossRef]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The Arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 2015, 53, 474–485. [Google Scholar] [CrossRef]
- Zheng, Y.; Wu, S.; Bai, Y.; Sun, H.; Jiao, C.; Guo, S.; Zhao, K.; Blanca, J.; Zhang, Z.; Huang, S.; et al. Cucurbit Genomics Database (CuGenDB): A central portal for comparative and functional genomics of cucurbit crops. Nucleic Acids Res. 2019, 47, D1128–D1136. [Google Scholar] [CrossRef]
- Duvick, J.; Fu, A.; Muppirala, U.; Sabharwal, M.; Wilkerson, M.D.; Lawrence, C.J.; Lushbough, C.; Brendel, V. PlantGDB: A resource for comparative plant genomics. Nucleic Acids Res. 2008, 36, D959–D965. [Google Scholar] [CrossRef]
- Fernandez-Pozo, N.; Menda, N.; Edwards, J.D.; Saha, S.; Tecle, I.Y.; Strickler, S.R.; Bombarely, A.; Fisher-York, T.; Pujar, A.; Foerster, H.; et al. The Sol Genomics Network (SGN)--from genotype to phenotype to breeding. Nucleic Acids Res. 2015, 43, D1036–D1041. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, M.R.; Cannon, E.K.; Portwood, J.L.; Harper, L.C.; Gardiner, J.M.; Schaeffer, M.L.; Andorf, C.M. A pan-genomic approach to genome databases using maize as a model system. BMC Plant Biol. 2021, 21, 385. [Google Scholar] [CrossRef] [PubMed]
- Sundell, D.; Mannapperuma, C.; Netotea, S.; Delhomme, N.; Lin, Y.C.; Sjodin, A.; Van de Peer, Y.; Jansson, S.; Hvidsten, T.R.; Street, N.R. The plant genome integrative explorer resource: PlantGenIE.org. New Phytol. 2015, 208, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.L.; Kohchi, T.; Yamato, K.T.; Jenkins, J.; Shu, S.Q.; Ishizaki, K.; Yamaoka, S.; Nishihama, R.; Nakamura, Y.; Berger, F.; et al. Insights into land plant evolution garnered from the genome. Cell 2017, 171, 287–304.e15. [Google Scholar] [CrossRef]
- Fernandez-Pozo, N.; Haas, F.B.; Meyberg, R.; Ullrich, K.K.; Hiss, M.; Perroud, P.F.; Hanke, S.; Kratz, V.; Powell, A.F.; Vesty, E.F.; et al. PEATmoss (Physcomitrella Expression Atlas Tool): A unified gene expression atlas for the model plant Physcomitrella patens. Plant J. 2020, 102, 165–177. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef]
- Loytynoja, A.; Goldman, N. An algorithm for progressive multiple alignment of sequences with insertions. Proc. Natl. Acad. Sci. USA 2005, 102, 10557–10562. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Julca, I.; Ferrari, C.; Flores-Tornero, M.; Proost, S.; Lindner, A.C.; Hackenberg, D.; Steinbachova, L.; Michaelidis, C.; Gomes Pereira, S.; Misra, C.S.; et al. Comparative transcriptomic analysis reveals conserved programmes underpinning organogenesis and reproduction in land plants. Nat. Plants 2021, 7, 1143–1159. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [CrossRef]
- Avni, R.; Nave, M.; Barad, O.; Baruch, K.; Twardziok, S.O.; Gundlach, H.; Hale, I.; Mascher, M.; Spannagl, M.; Wiebe, K.; et al. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 2017, 357, 93–97. [Google Scholar] [CrossRef]
- Luo, M.C.; Gu, Y.Q.; Puiu, D.; Wang, H.; Twardziok, S.O.; Deal, K.R.; Huo, N.; Zhu, T.; Wang, L.; Wang, Y.; et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 2017, 551, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.Q.; Ma, B.; Shi, X.; Liu, H.; Dong, L.; Sun, H.; Cao, Y.; Gao, Q.; Zheng, S.; Li, Y.; et al. Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 2018, 557, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Appels, R.; Eversole, K.; Feuillet, C.; Keller, B.; Rogers, J.; Stein, N.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; Poland, J.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef]
- Lescot, M.; Dehais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouze, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Wang, J.; Fu, W.; Wang, R.; Hu, D.; Cheng, H.; Zhao, J.; Jiang, Y.; Kang, Z. WGVD: An integrated web-database for wheat genome variation and selective signatures. Database 2020, 2020, baaa090. [Google Scholar] [CrossRef]
- Chen, Y.M.; Song, W.J.; Xie, X.M.; Wang, Z.H.; Guan, P.F.; Peng, H.R.; Jiao, Y.N.; Ni, Z.F.; Sun, Q.X.; Guo, W.L. A collinearity-incorporating homology inference strategy for connecting emerging assemblies in the Triticeae tribe as a pilot practice in the plant pangenomic era. Mol. Plant 2020, 13, 1694–1708. [Google Scholar] [CrossRef]
- Ma, S.; Wang, M.; Wu, J.; Guo, W.; Chen, Y.; Li, G.; Wang, Y.; Shi, W.; Xia, G.; Fu, D.; et al. WheatOmics: A platform combining multiple omics data to accelerate functional genomics studies in wheat. Mol. Plant 2021, 14, 1965–1968. [Google Scholar] [CrossRef]
- Schrodinger, LLC. The AxPyMOL Molecular Graphics Plugin for Microsoft PowerPoint, Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Transcript ID | Gene Name | Protein Length | Mw | pI | GRAVY |
|---|---|---|---|---|---|---|
| T. urartu | TuG1812S0000898200.01.P01 | Tu_TRM6_S1 1 | 351 | 37,991.54 | 7.66 | −0.144 |
| T. urartu | TuG1812S0002603300.01.P01 | Tu_TRM6_S2 | 511 | 54,931.29 | 5.43 | −0.273 |
| A. tauschii | AET2Gv20006900.9 | Aet_TRM6_2D | 480 | 51,766.75 | 5.5 | −0.243 |
| A. tauschii | AET3Gv20046100.2 | Aet_TRM6_3D | 475 | 50,921.88 | 6.12 | −0.235 |
| T. dicoccoides | TRIDC2AG000170.1 | Td_TRM6_2A | 294 | 31,273.83 | 4.56 | −0.458 |
| T. dicoccoides | TRIDC3AG000690.1 | Td_TRM6_3A | 356 | 37,786.76 | 5.18 | −0.242 |
| T. dicoccoides | TRIDC3BG003460.1 | Td_TRM6_3B | 476 | 51,160.16 | 8.89 | −0.364 |
| T. aestivum | TraesCS2A02G004800.1 | Ta_TRM6_2A | 507 | 54,463.62 | 5.29 | −0.286 |
| T. aestivum | TraesCS2D02G004000.1 | Ta_TRM6_2D | 483 | 51,921.91 | 5.61 | −0.263 |
| T. aestivum | TraesCS3A02G024400.1 | Ta_TRM6_3A | 473 | 50,831.81 | 5.96 | −0.208 |
| T. aestivum | TraesCS3B02G020400.1 | Ta_TRM6_3B | 474 | 50,670.67 | 5.81 | −0.194 |
| T. aestivum | TraesCS3D02G021800.1 | Ta_TRM6_3D | 481 | 51,585.71 | 5.96 | −0.197 |
| T. aestivum | TraesCSU02G012500.1 | Ta_TRM6_U 2 | 517 | 55,236.48 | 5.42 | −0.269 |
| T. urartu | TuG1812G0200003197.01.P01 | Tu_TRM61_2A | 335 | 35,973.76 | 7.61 | −0.106 |
| A. tauschii | AET2Gv20616500.1 | Aet_TRM61_2D | 336 | 35,939.64 | 7.13 | −0.124 |
| T. dicoccoides | TRIDC2AG039430.1 | Td_TRM61_2A | 336 | 35,982.77 | 7.62 | −0.105 |
| T. dicoccoides | TRIDC2BG042450.1 | Td_TRM61_2B | 336 | 35,925.65 | 7.64 | −0.113 |
| T. aestivum | TraesCS2A02G273600.1 | Ta_TRM61_2A | 335 | 35,982.77 | 7.62 | −0.105 |
| T. aestivum | TraesCS2B02G291400.1 | Ta_TRM61_2B | 335 | 35,939.68 | 7.64 | −0.114 |
| T. aestivum | TraesCS2D02G272600.1 | Ta_TRM61_2D | 335 | 35,939.64 | 7.13 | −0.124 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yue, W.; Chen, T.; Liu, S.; Shi, X. Evolutionary Dynamics of Plant TRM6/TRM61 Complexes. Plants 2025, 14, 1778. https://doi.org/10.3390/plants14121778
Yue W, Chen T, Liu S, Shi X. Evolutionary Dynamics of Plant TRM6/TRM61 Complexes. Plants. 2025; 14(12):1778. https://doi.org/10.3390/plants14121778
Chicago/Turabian StyleYue, Wenjie, Tong Chen, Shuyi Liu, and Xiaowen Shi. 2025. "Evolutionary Dynamics of Plant TRM6/TRM61 Complexes" Plants 14, no. 12: 1778. https://doi.org/10.3390/plants14121778
APA StyleYue, W., Chen, T., Liu, S., & Shi, X. (2025). Evolutionary Dynamics of Plant TRM6/TRM61 Complexes. Plants, 14(12), 1778. https://doi.org/10.3390/plants14121778