
Genetic Mapping of QTL Associated with 100-Kernel Weight Using a DH Population in Maize

 ,
,  ,
, 
Abstract
1. Introduction
2. Results
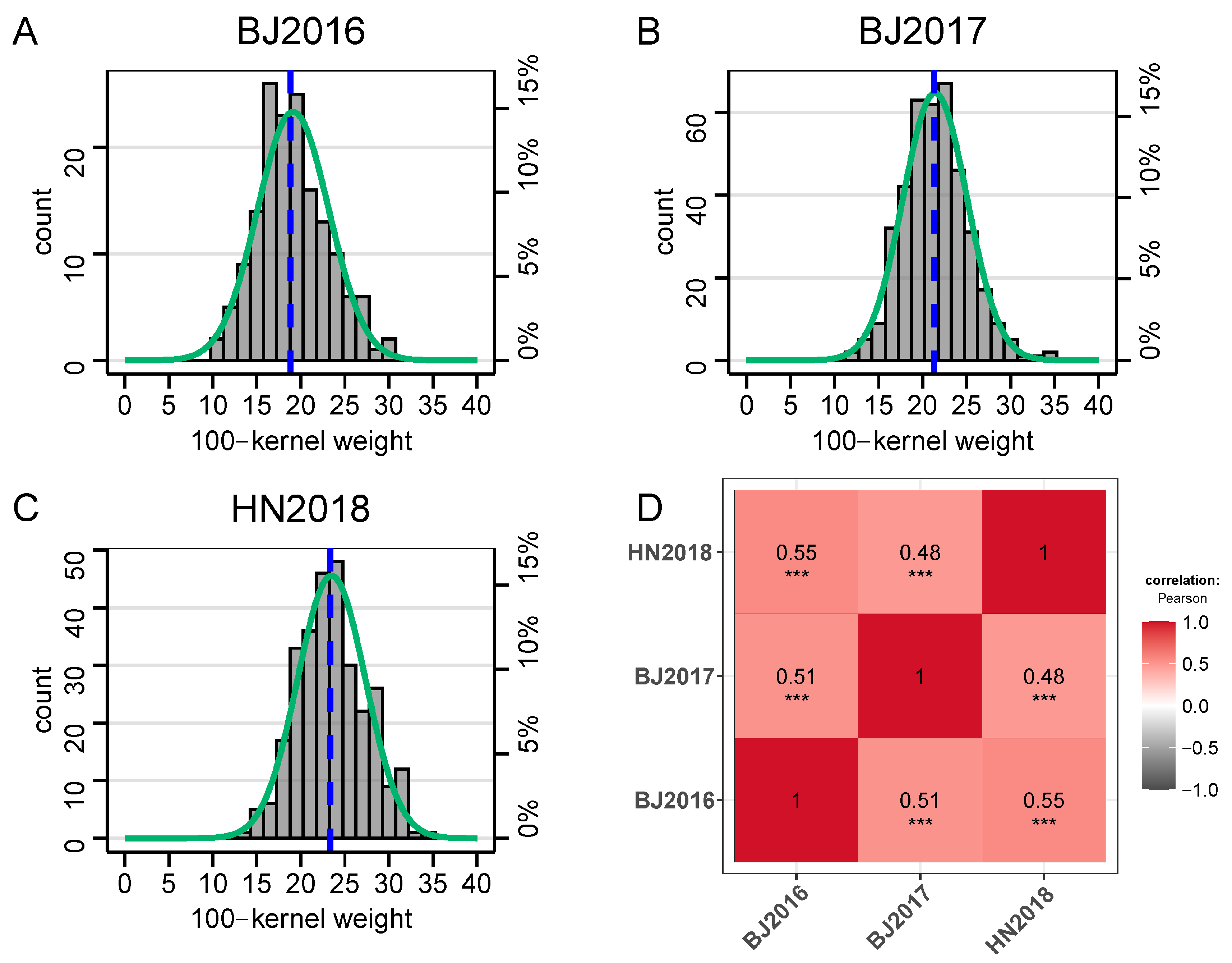
2.1. Phenotypic Variations of 100-Kernel Weight in DH Population
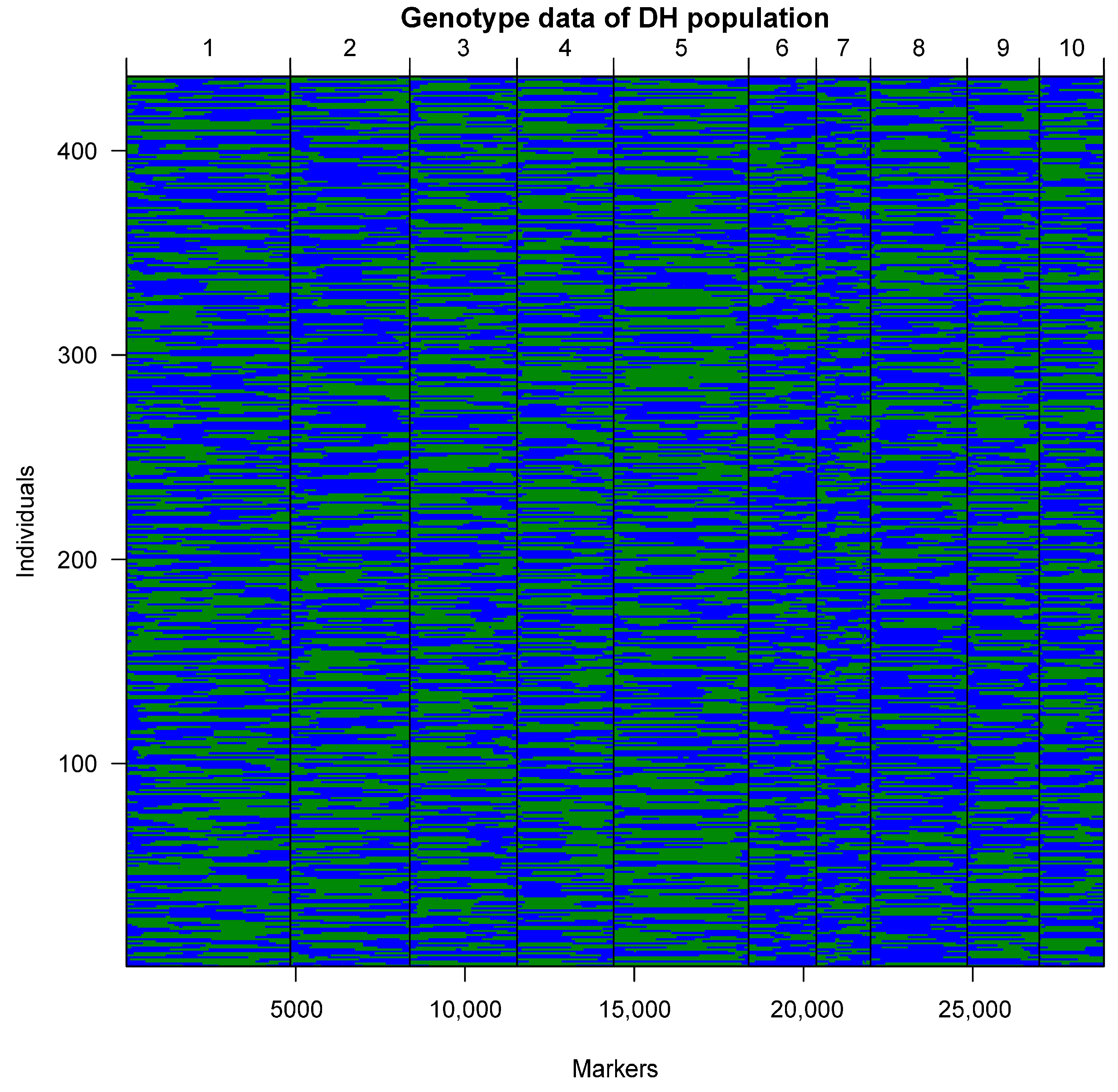
2.2. Construction of the Genetic Linkage Map
2.3. QTL Mapping and Analysis for 100-Kernel Weight
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Phenotyping
4.2. Genetic Linkage Map Construction
4.3. QTL Mapping and Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DH | Double Haploid |
| QTL | Quantitative Trait Loci |
| MP3RNA-seq | Massively Parallel 3′ end RNA sequencing |
| SNP | Single Nucleotide Polymorphism |
| HKW | 100-Kernel Weight |
| RIL | recombination inbred line |
| BC | BackCross |
| NAM panel | Nested Association Mapping panel |
| MAGIC | Multiparent Advanced Generation Inter-Cross |
| GWAS | Genome-Wide Association Studies |
| CV | Coefficient of Variation |
| LOD | Logarithm of the Odds |
| PVE | Phenotypic Variance Explained |
| BLUP | Best Linear Unbiased Prediction |
| CIM | Composite Interval Mapping |
| BJ2016 | Beijing2016 |
| BJ2017 | Beijing2017 |
| HN2018 | Hainan2018 |
| MARS | Marker-Assisted Recurrent Selection |
| GE | Genome Editing |
| KW | Kernel Weight |
| KN | Kernel Number |
References
- Wang, B.; Lin, Z.; Li, X.; Zhao, Y.; Zhao, B.; Wu, G.; Ma, X.; Wang, H.; Xie, Y.; Li, Q.; et al. Genome-wide selection and genetic improvement during modern maize breeding. Nat. Genet. 2020, 52, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Z.; Tan, K.; Huang, W.; Shi, J.; Li, T.; Hu, J.; Wang, K.; Wang, C.; Xin, B.; et al. A complete telomere-to-telomere assembly of the maize genome. Nat. Genet. 2023, 55, 1221–1231. [Google Scholar] [CrossRef]
- Gao, X.; Mo, W.; Shi, J.; Song, N.; Liang, P.; Chen, J.; Shi, Y.; Guo, W.; Li, X.; Yang, X.; et al. HITAC-seq enables high-throughput cost-effective sequencing of plasmids and DNA fragments with identity. J. Genet. Genom. 2021, 48, 671–680. [Google Scholar] [CrossRef]
- Gao, Q.; Yue, G.; Li, W.; Wang, J.; Xu, J.; Yin, Y. Recent progress using high-throughput sequencing technologies in plant molecular breeding. J. Integr. Plant Biol. 2012, 54, 215–227. [Google Scholar] [CrossRef]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Eshenour, K.; Hotto, A.; Michel, E.J.S.; Oh, Z.G.; Stern, D.B. Transgenic expression of Rubisco accumulation factor2 and Rubisco subunits increases photosynthesis and growth in maize. J. Exp. Bot. 2024, 75, 4024–4037. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Jiang, Y.; Sun, M.; Wang, Y.; Zhang, L.; Zeng, X.; Wang, Z.; Wang, Z.; Liu, Y.; Gan, Y.; et al. Bi-parental graph strategy to represent and analyze hybrid plant genomes. Plant Physiol. 2024, 196, 1284–1297. [Google Scholar] [CrossRef]
- Cagnola, J.I.; D’Andrea, K.E.; Rotili, D.H.; Mercau, J.L.; Ploschuk, E.L.; Maddonni, G.A.; Otegui, M.E.; Casal, J.J. Eco-physiology of maize crops under combined stresses. Plant J. 2024, 117, 1856–1872. [Google Scholar] [CrossRef]
- Tian, J.; Wang, C.; Chen, F.; Qin, W.; Yang, H.; Zhao, S.; Xia, J.; Du, X.; Zhu, Y.; Wu, L.; et al. Maize smart-canopy architecture enhances yield at high densities. Nature 2024, 632, 576–584. [Google Scholar] [CrossRef]
- Wang, B.; Liu, H.; Liu, Z.; Dong, X.; Guo, J.; Li, W.; Chen, J.; Gao, C.; Zhu, Y.; Zheng, X.; et al. Identification of minor effect QTLs for plant architecture related traits using super high density genotyping and large recombinant inbred population in maize (Zea mays). BMC Plant Biol. 2018, 18, 17. [Google Scholar] [CrossRef]
- Hamwieh, A.; Xu, D.H. Conserved salt tolerance quantitative trait locus (QTL) in wild and cultivated soybeans. Breed. Sci. 2008, 58, 355–359. [Google Scholar] [CrossRef]
- Jia, Y.H.; Sun, X.W.; Sun, J.L.; Pan, Z.E.; Wang, X.W.; He, S.P.; Xiao, S.H.; Shi, W.J.; Zhou, Z.L.; Pang, B.Y.; et al. Association Mapping for Epistasis and Environmental Interaction of Yield Traits in 323 Cotton Cultivars under 9 Different Environments. PLoS ONE 2014, 9, e95882. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, H.; Zhu, Y.; Huang, X.; Li, S.; Wu, X.; Zhao, Y.; Bao, Z.; Qin, L.; Jin, Y.; et al. THP9 enhances seed protein content and nitrogen-use efficiency in maize. Nature 2022, 612, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Barriere, Y.; Mechin, V.; Lefevre, B.; Maltese, S. QTLs for agronomic and cell wall traits in a maize RIL progeny derived from a cross between an old Minnesota13 line and a modern Iodent line. Theor. Appl. Genet. 2012, 125, 531–549. [Google Scholar] [CrossRef]
- Dell’Acqua, M.; Gatti, D.M.; Pea, G.; Cattonaro, F.; Coppens, F.; Magris, G.; Hlaing, A.L.; Aung, H.H.; Nelissen, H.; Baute, J.; et al. Genetic properties of the MAGIC maize population: A new platform for high definition QTL mapping in Zea mays. Genome Biol. 2015, 16, 167. [Google Scholar] [CrossRef]
- Kahler, A.L.; Wehrhahn, C.F. Associations between quantitative traits and enzyme loci in the F2 population of a maize hybrid. Theor. Appl. Genet. 1986, 72, 15–26. [Google Scholar] [CrossRef]
- McMullen, M.D.; Kresovich, S.; Villeda, H.S.; Bradbury, P.; Li, H.; Sun, Q.; Flint-Garcia, S.; Thornsberry, J.; Acharya, C.; Bottoms, C.; et al. Genetic properties of the maize nested association mapping population. Science 2009, 325, 737–740. [Google Scholar] [CrossRef]
- Yang, D.E.; Jin, D.M.; Wang, B.; Zhang, D.S.; Nguyen, H.T.; Zhang, C.L.; Chen, S.J. Characterization and mapping of Rpi1, a gene that confers dominant resistance to stalk rot in maize. Mol. Genet. Genom. 2005, 274, 229–234. [Google Scholar] [CrossRef]
- Li, K.; Tassinari, A.; Giuliani, S.; Rosignoli, S.; Urbany, C.; Tuberosa, R.; Salvi, S. QTL mapping identifies novel major loci for kernel row number-associated ear fasciation, ear prolificacy and tillering in maize (Zea mays L.). Front. Plant Sci. 2022, 13, 1017983. [Google Scholar] [CrossRef]
- Sa, K.J.; Choi, I.Y.; Park, J.Y.; Choi, J.K.; Ryu, S.H.; Lee, J.K. Mapping of QTL for agronomic traits using high-density SNPs with an RIL population in maize. Genes. Genom. 2021, 43, 1403–1411. [Google Scholar] [CrossRef]
- Jain, S.M.; Brar, D.S. QTL Analysis in Plant Breeding. In Molecular Techniques in Crop Improvement; Springer Netherlands: Berlin/Heidelberg, Germany, 2009; pp. 3–21. [Google Scholar]
- Chen, L.; Li, Y.X.; Li, C.; Wu, X.; Qin, W.; Li, X.; Jiao, F.; Zhang, X.; Zhang, D.; Shi, Y.; et al. Fine-mapping of qGW4.05, a major QTL for kernel weight and size in maize. BMC Plant Biol. 2016, 16, 81. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, Z.; Zhang, X.; Liu, H.; Zhou, L.; Zhong, S.; Li, Y.; Zhu, C.; Lin, Z. krn1, a major quantitative trait locus for kernel row number in maize. New Phytol. 2019, 223, 1634–1646. [Google Scholar] [CrossRef]
- Li, W.; Bai, Q.; Zhan, W.; Ma, C.; Wang, S.; Feng, Y.; Zhang, M.; Zhu, Y.; Cheng, M.; Xi, Z. Fine mapping and candidate gene analysis of qhkw5-3, a major QTL for kernel weight in maize. Theor. Appl. Genet. 2019, 132, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Dong, Y.; Shi, Q.; Zhang, L.; Chen, H.; Hu, C.; Li, Y. Verification and fine mapping of qGW1.05, a major QTL for grain weight in maize (Zea mays L.). Mol. Genet. Genom. 2017, 292, 871–881. [Google Scholar] [CrossRef]
- Hoshino, T.; Kawashita, N.; Takagi, Y.; Anai, T. Molecular characterization and marker development of mid-oleic-acid mutant M23 for the development of high-oleic cultivars of soybean. Plant Breed. 2011, 130, 544–550. [Google Scholar] [CrossRef]
- Multani, D.S.; Briggs, S.P.; Chamberlin, M.A.; Blakeslee, J.J.; Murphy, A.S.; Johal, G.S. Loss of an MDR transporter in compact stalks of maize br2 and sorghum dw3 mutants. Science 2003, 302, 81–84. [Google Scholar] [CrossRef]
- Tian, J.; Wang, C.; Xia, J.; Wu, L.; Xu, G.; Wu, W.; Li, D.; Qin, W.; Han, X.; Chen, Q.; et al. Teosinte ligule allele narrows plant architecture and enhances high-density maize yields. Science 2019, 365, 658–664. [Google Scholar] [CrossRef]
- Hui, G.Q.; Wen, G.Q.; Liu, X.H.; Yang, H.P.; Luo, Q.; Song, H.X.; Wen, L.; Sun, Y.; Zhang, H.M. Quantitative trait locus analysis for kernel width using maize recombinant inbred lines. Genet. Mol. Res. 2015, 14, 14496–14502. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, X.; Yi, F.; Gao, X.; Song, W.; Zhao, H.; Lai, J. MP3RNA-seq: Massively parallel 3′ end RNA sequencing for high-throughput gene expression profiling and genotyping. J. Integr. Plant Biol. 2021, 63, 1227–1239. [Google Scholar] [CrossRef]
- Gao, R.X.; Hu, M.J.; Zhao, H.M.; Lai, J.S.; Song, W.B. Genetic dissection of ear-related traits using immortalized F2 population in maize. J. Integr. Agric. 2022, 21, 2492–2507. [Google Scholar] [CrossRef]
- Li, H.; Yang, Q.; Fan, N.; Zhang, M.; Zhai, H.; Ni, Z.; Zhang, Y. Quantitative trait locus analysis of heterosis for plant height and ear height in an elite maize hybrid zhengdan 958 by design III. BMC Genet. 2017, 18, 36. [Google Scholar] [CrossRef]
- Ma, J.; Tu, Y.; Zhu, J.; Luo, W.; Liu, H.; Li, C.; Li, S.; Liu, J.; Ding, P.; Habib, A.; et al. Flag leaf size and posture of bread wheat: Genetic dissection, QTL validation and their relationships with yield-related traits. Theor. Appl. Genet. 2020, 133, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zeng, B.; Zhang, M.; Xie, S.; Wang, G.; Hauck, A.; Lai, J. Dynamic transcriptome landscape of maize embryo and endosperm development. Plant Physiol. 2014, 166, 252–264. [Google Scholar] [CrossRef]
- Yu, J.; Holland, J.B.; McMullen, M.D.; Buckler, E.S. Genetic design and statistical power of nested association mapping in maize. Genetics 2008, 178, 539–551. [Google Scholar] [CrossRef]
- Zhu, Y.; Song, B.; Guo, Y.; Wang, B.; Xu, C.; Zhu, H.; E, L.; Lai, J.; Song, W.; Zhao, H. QTL Analysis Reveals Conserved and Differential Genetic Regulation of Maize Lateral Angles above the Ear. Plants 2023, 12, 680. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.K.; Sa, K.J.; Park, D.H.; Lim, S.E.; Ryu, S.H.; Park, J.Y.; Park, K.J.; Rhee, H.I.; Lee, M.; Lee, J.K. Construction of genetic linkage map and identification of QTLs related to agronomic traits in DH population of maize (Zea mays L.) using SSR markers. Genes Genom. 2019, 41, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liang, T.; Chen, M.; Zhang, Y.; Wang, T.; Lin, H.; Rong, T.; Zou, C.; Liu, P.; Lee, M.; et al. Genetic dissection of stalk lodging-related traits using an IBM Syn10 DH population in maize across three environments (Zea mays L.). Mol. Genet. Genom. 2019, 294, 1277–1288. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Z.; Chen, Q.; Qu, Y.; Tang, J.; Lubberstedt, T.; Li, H. Mapping of QTL for Grain Yield Components Based on a DH Population in Maize. Sci. Rep. 2020, 10, 7086. [Google Scholar] [CrossRef]
- Kchouk, M.; Gibrat, J.F.; Elloumi, M. Generations of Sequencing Technologies: From First to Next Generation. Biol. Med. 2017, 9, 3. [Google Scholar] [CrossRef]
- Lai, J.; Li, R.; Xu, X.; Jin, W.; Xu, M.; Zhao, H.; Xiang, Z.; Song, W.; Ying, K.; Zhang, M.; et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 2010, 42, 1027–1030. [Google Scholar] [CrossRef]
- Wang, B.; Hou, M.; Shi, J.; Ku, L.; Song, W.; Li, C.; Ning, Q.; Li, X.; Li, C.; Zhao, B.; et al. De novo genome assembly and analyses of 12 founder inbred lines provide insights into maize heterosis. Nat. Genet. 2023, 55, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.J.; Wang, X.; Xiao, Y.; Luo, J.; Qiao, F.; Yang, W.; Zhang, R.; Meng, Y.; Sun, J.; Yan, S.; et al. CUBIC: An atlas of genetic architecture promises directed maize improvement. Genome Biol. 2020, 21, 20. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Chen, M.; Zhang, Q.; Gong, J.; Liu, J.; Yong, K.; Wang, Q.; Fan, J.; Chen, S.; Hua, H.; et al. Genomic investigation of 18,421 lines reveals the genetic architecture of rice. Science 2024, 385, eadm8762. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhuang, Z.; Bian, J.; Tang, R.; Ren, Z.; Peng, Y. Candidate Gene for Kernel-Related Traits in Maize Revealed by a Combination of GWAS and Meta-QTL Analyses. Plants 2025, 14, 959. [Google Scholar] [CrossRef]
- Wang, M.; Cheng, D.; Ren, H.; Li, H.; Wang, R.; Dong, C.; Chang, J. Genome-Wide Association Analysis Identifies Loci and Candidate Genes for 100-Kernel Weight in Maize. Agronomy 2024, 14, 2954. [Google Scholar] [CrossRef]
- Ramya, P.; Chaubal, A.; Kulkarni, K.; Gupta, L.; Kadoo, N.; Dhaliwal, H.S.; Chhuneja, P.; Lagu, M.; Gupta, V. QTL mapping of 1000-kernel weight, kernel length, and kernel width in bread wheat (Triticum aestivum L.). J. Appl. Genet. 2010, 51, 421–429. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.X.; Chen, L.; Wu, X.; Qin, W.W.; Song, Y.C.; Zhang, D.F.; Wang, T.Y.; Li, Y.; Shi, Y.S. Fine mapping of qKW7, a major QTL for kernel weight and kernel width in maize, confirmed by the combined analytic approaches of linkage and association analysis. Euphytica 2016, 210, 221–232. [Google Scholar] [CrossRef]
- Yang, N.; Liu, J.; Gao, Q.; Gui, S.T.; Chen, L.; Yang, L.F.; Huang, J.; Deng, T.Q.; Luo, J.Y.; He, L.J.; et al. Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement. Nat. Genet. 2019, 51, 1052–1059. [Google Scholar] [CrossRef]
- Hufford, M.B.; Xu, X.; van Heerwaarden, J.; Pyhäjärvi, T.; Chia, J.-M.; Cartwright, R.A.; Elshire, R.J.; Glaubitz, J.C.; Guill, K.E.; Kaeppler, S.M.; et al. Comparative population genomics of maize domestication and improvement. Nat. Genet. 2012, 44, 808–811. [Google Scholar] [CrossRef]
- Fernández, J.A.; Messina, C.D.; Salinas, A.; Prasad, P.V.V.; Nippert, J.B.; Ciampitti, I.A. Kernel weight contribution to yield genetic gain of maize: A global review and US case studies. J. Exp. Bot. 2022, 73, 3597–3609. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, X.; Fu, J.; Wang, H.; Wang, J.; Huang, C.; Prasanna, B.M.; Olsen, M.S.; Wang, G.; Zhang, A. Enhancing Genetic Gain through Genomic Selection: From Livestock to Plants. Plant Commun. 2020, 1, 100005. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yi, Q.; Hou, X.; Hu, Y.; Huang, Y. Identification of quantitative trait loci for kernel-related traits and the heterosis for these traits in maize (Zea mays L.). Mol. Genet. Genomics 2020, 295, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.B. Theoretical basis for separation of multiple linked gene effects in mapping quantitative trait loci. Proc. Natl. Acad. Sci. USA 1993, 90, 10972–10976. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Wang, B.; Hauck, A.L.; Dong, X.; Li, J.; Lai, J. Genetic dissection of maize seedling root system architecture traits using an ultra-high density bin-map and a recombinant inbred line population. J. Integr. Plant Biol. 2016, 58, 266–279. [Google Scholar] [CrossRef]
- Tang, J.; Yan, J.; Ma, X.; Teng, W.; Wu, W. Dissection of the genetic basis of heterosis in an elite maize hybrid by QTL mapping in an immortalized F2 population. Theor. Appl. Genet. 2010, 120, 333–340. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Env. a | Mean ± SD (g) | Range (g) | Kurtosis | Skew | CV b (%) | h2 c (%) | |
|---|---|---|---|---|---|---|---|
| Population | BJ2016 | 19.12 ± 4.05 | 10.45–30.52 | 0.03 | 0.41 | 21.17 | 79.70 |
| BJ2017 | 21.40 ± 3.64 | 11.60–34.50 | 0.49 | 0.36 | 17.02 | ||
| HN2018 | 23.47 ± 3.85 | 13.86–33.87 | −0.38 | 0.17 | 16.39 | ||
| BLUP | 21.07 ± 1.88 | 16.06–28.83 | 0.86 | 0.37 | 8.91 | ||
| Overall | 21.49 ± 3.53 | 10.45–34.50 | 0.74 | 0.35 | 16.43 | ||
| C7-2 | BJ2016 | 26.31 ± 3.22 | |||||
| BJ2017 | 29.10 ± 2.67 | ||||||
| HN2018 | 28.71 ± 3.00 | ||||||
| PHBA6 | BJ2016 | 18.53 ± 4.04 | |||||
| BJ2017 | 20.91 ± 2.51 | ||||||
| HN2018 | 21.12 ± 3.62 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Li, H.; Chen, J.; Zhang, X.; Wang, B.; Zhi, S.; Guan, H.; Song, W.; Lai, J.; Zhao, H.; et al. Genetic Mapping of QTL Associated with 100-Kernel Weight Using a DH Population in Maize. Plants 2025, 14, 1737. https://doi.org/10.3390/plants14121737
Li H, Li H, Chen J, Zhang X, Wang B, Zhi S, Guan H, Song W, Lai J, Zhao H, et al. Genetic Mapping of QTL Associated with 100-Kernel Weight Using a DH Population in Maize. Plants. 2025; 14(12):1737. https://doi.org/10.3390/plants14121737
Chicago/Turabian StyleLi, Huawei, Hao Li, Jian Chen, Xiangbo Zhang, Baobao Wang, Shujun Zhi, Haiying Guan, Weibin Song, Jinsheng Lai, Haiming Zhao, and et al. 2025. "Genetic Mapping of QTL Associated with 100-Kernel Weight Using a DH Population in Maize" Plants 14, no. 12: 1737. https://doi.org/10.3390/plants14121737
APA StyleLi, H., Li, H., Chen, J., Zhang, X., Wang, B., Zhi, S., Guan, H., Song, W., Lai, J., Zhao, H., & Gao, R. (2025). Genetic Mapping of QTL Associated with 100-Kernel Weight Using a DH Population in Maize. Plants, 14(12), 1737. https://doi.org/10.3390/plants14121737