

Unraveling the Nectar Secretion Pathway and Floral-Specific Expression of SWEET and CWIV Genes in Five Dandelion Species Through RNA Sequencing



Abstract
1. Introduction
2. Results
2.1. Assembly and Functional Annotation
2.2. Gene Ontology (GO) Terms Associated with Sucrose Biosynthesis in Taraxacum
2.3. KEGG Annotation
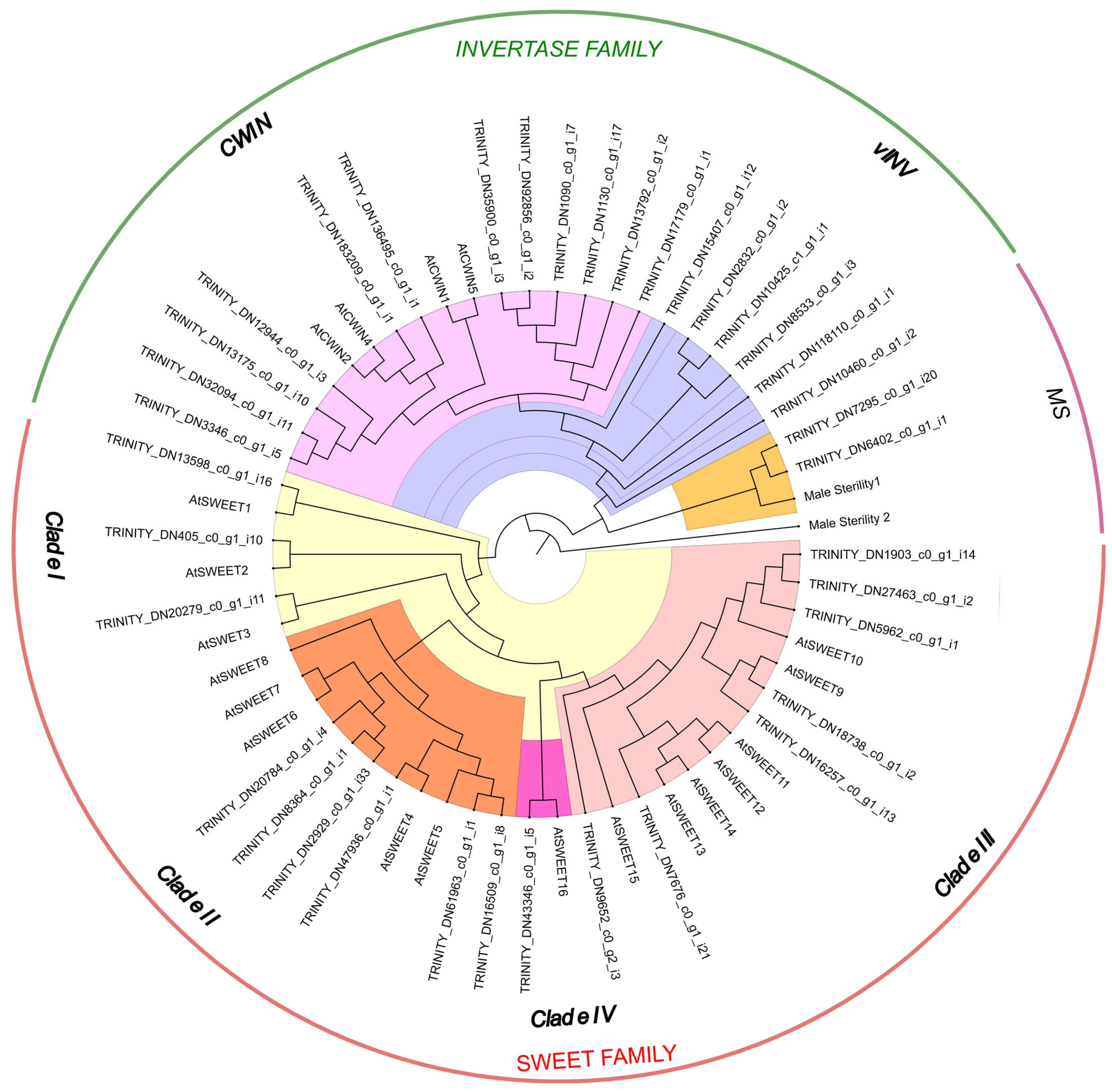
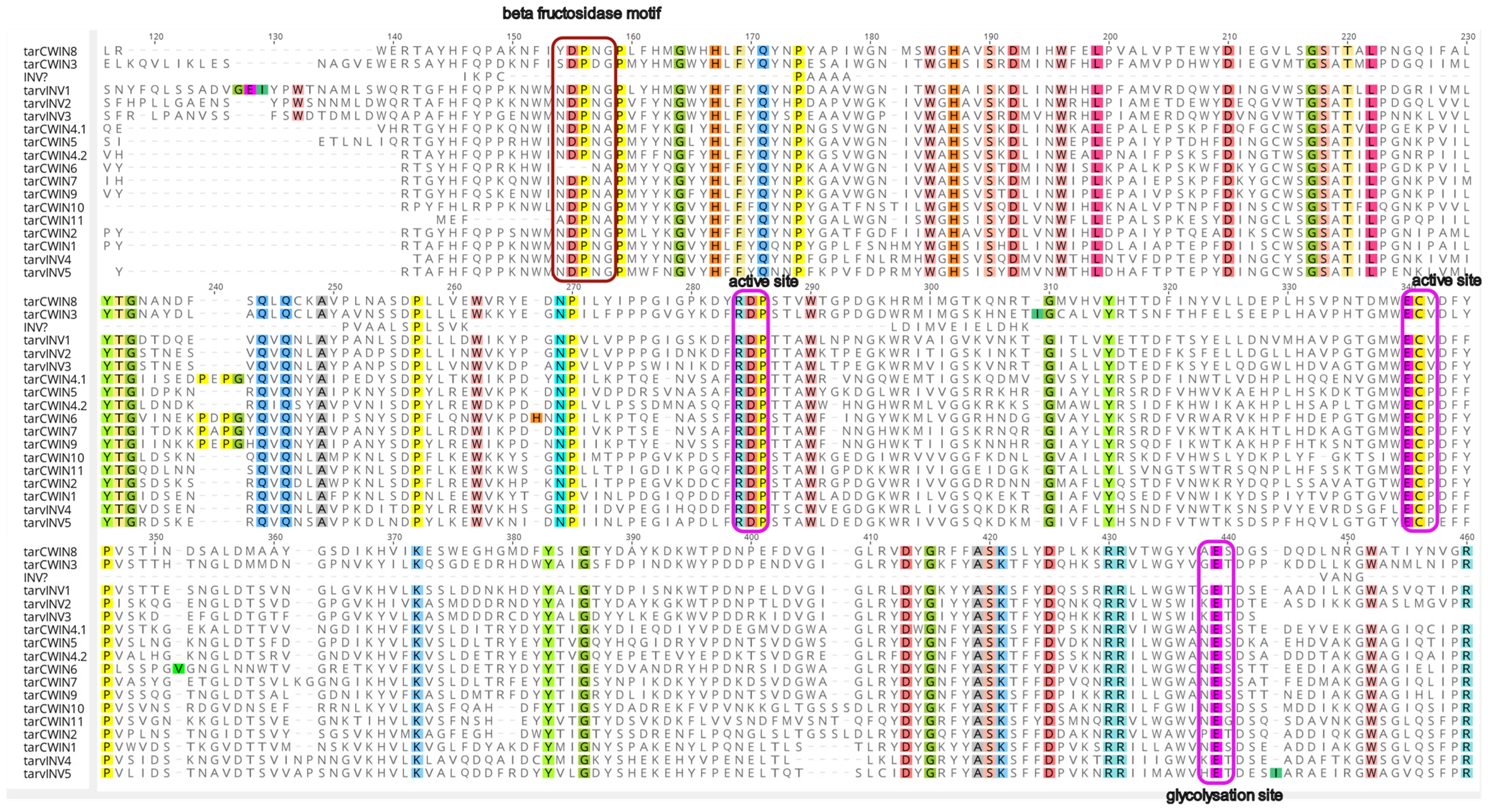
2.4. SWEET/CWIN/MS Genes and Their Phylogeny
- Clade I (SWEET1–3) and Clade II (SWEET4–8) were associated with glucose transport.
- Clade III (SWEET9–15) participated in sucrose transport.
- Clade IV (SWEET16) was linked to fructose transport.
2.5. DEG Analysis of SWEET/CWIN/MS Genes in Taraxacum Flowers
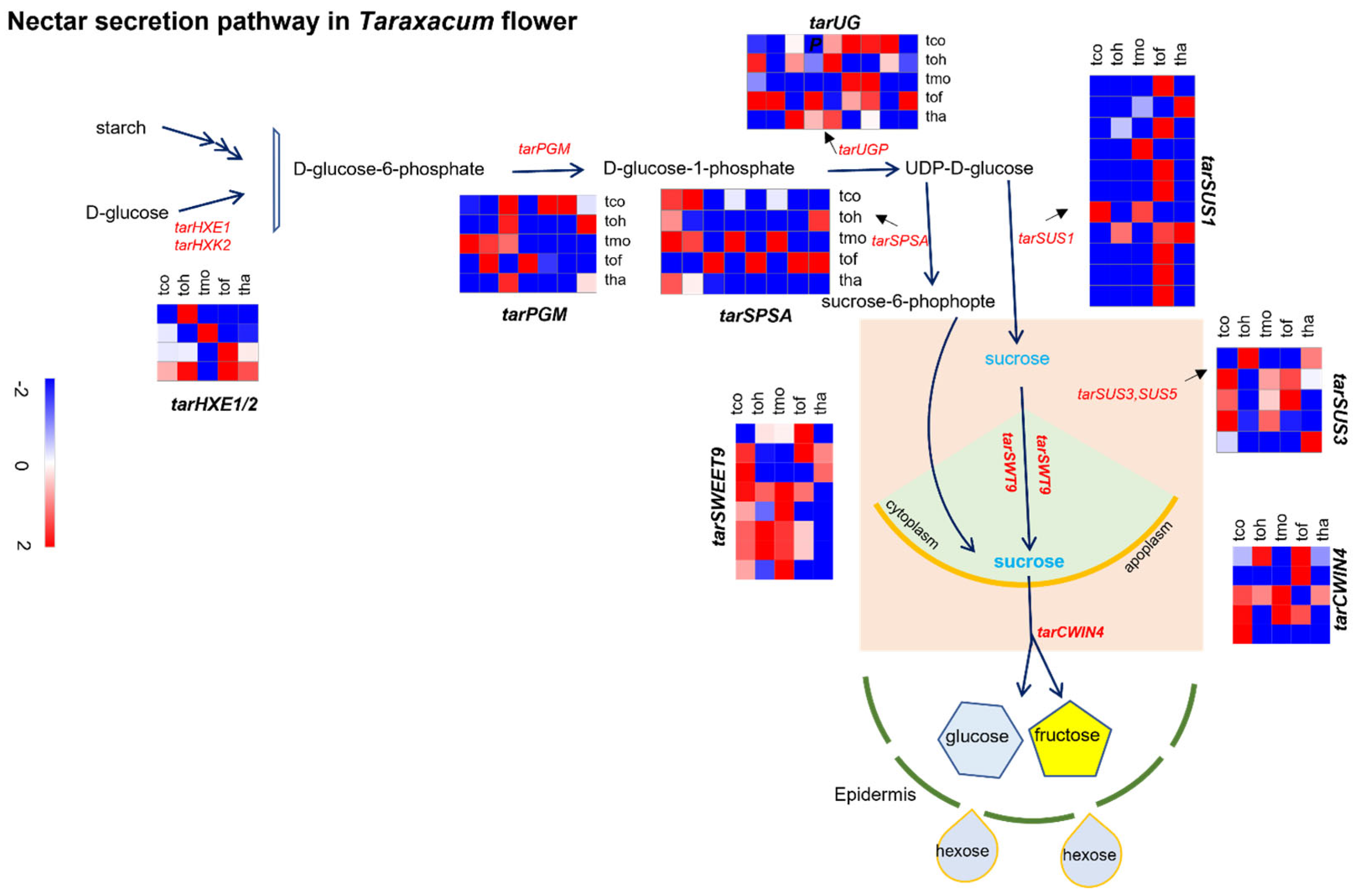
2.6. Analysis of Nectar Secretion Pathway and Associated Gene Expressions in Taraxacum Flowers
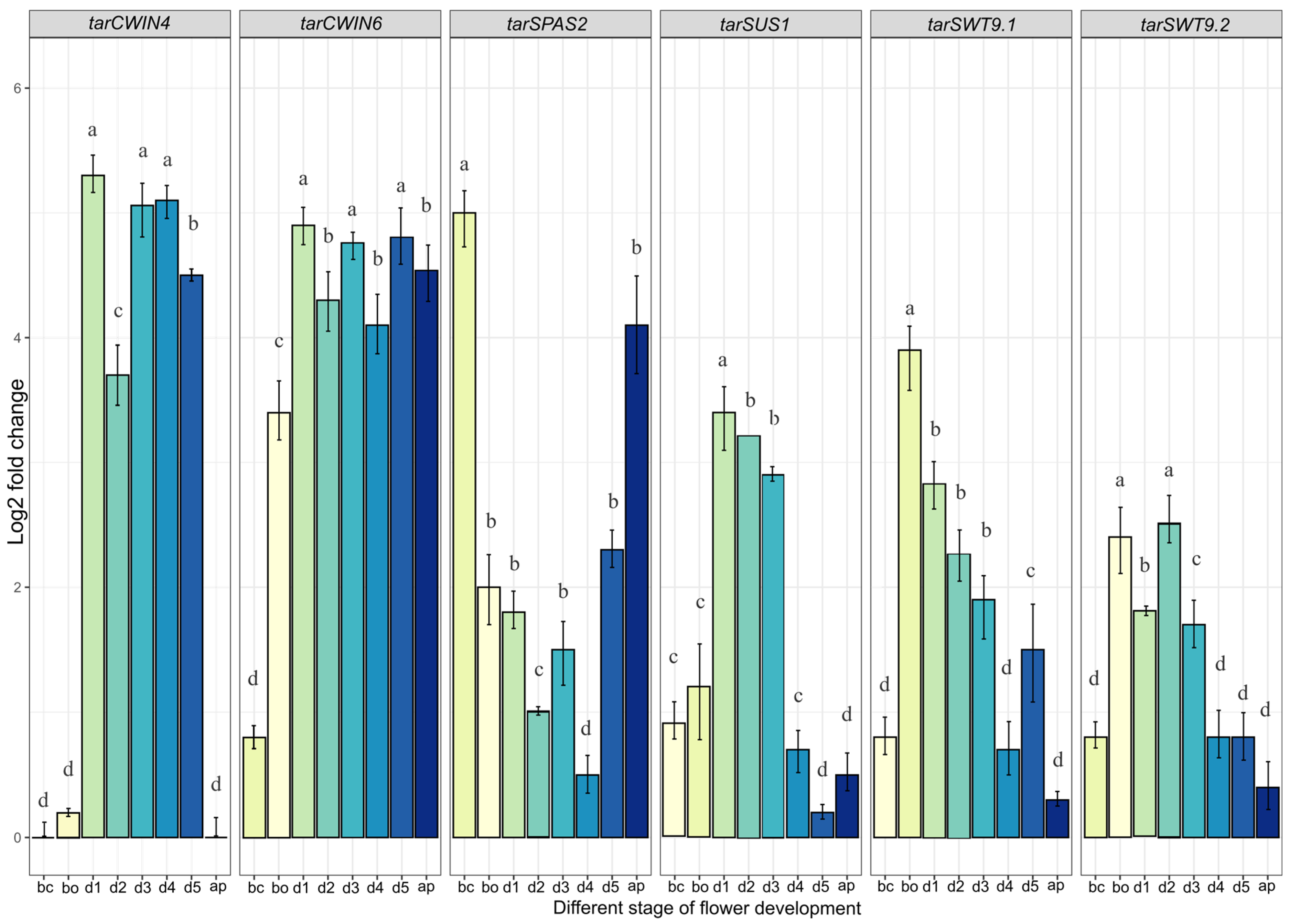
2.7. Quantification of Genes Involved in the Nectar Secretion Pathway in T. officinale
3. Discussion
4. Materials and Methods
4.1. Plant Materials, RNA Sequencing, and Assembly
4.2. Gene Annotation
4.3. Differential Gene Expression Analysis
4.4. Quantitative PCR Expression Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Baker, H.G.; Baker, I. Amino-acids in Nectar and their Evolutionary Significance. Nature 1973, 241, 543–545. [Google Scholar] [CrossRef]
- Gottsberger, G.; Schrauwen, J.; Linskens, H. Amino acids and sugars in nectar, and their putative evolutionary significance. Plant Syst. Evol. 1984, 145, 55–77. [Google Scholar] [CrossRef]
- Brandenburg, A.; Dell’Olivo, A.; Bshary, R.; Kuhlemeier, C. The sweetest thing Advances in nectar research. Curr. Opin. Plant Biol. 2009, 12, 486–490. [Google Scholar] [CrossRef] [PubMed]
- De la Barrera, E.; Nobel, P.S. Nectar: Properties, floral aspects, and speculations on origin. Trends Plant Sci. 2004, 9, 65–69. [Google Scholar] [CrossRef]
- González-Teuber, M.; Heil, M. Nectar chemistry is tailored for both attraction of mutualists and protection from exploiters. Plant Signal. Behav. 2009, 4, 809–813. [Google Scholar] [CrossRef]
- Morris, W.F.; Mangel, M.; Adler, F.R. Mechanisms of pollen deposition by insect pollinators. Evol. Ecol. 1995, 9, 304–317. [Google Scholar] [CrossRef]
- Baker, H.; Baker, I. Chemical constituents of nectar in relation to pollination mechanisms and phylogeny. In Biochemical Aspects of Evolutionary Biology; University of Chicago Press: Chicago, IL, USA, 1983. [Google Scholar]
- Baker, H.G.; Baker, I. The occurrence and significance of amino acids in floral nectar. Plant Syst. Evol. 1986, 151, 175–186. [Google Scholar] [CrossRef]
- Vesprini, J.L.; Nepi, M.; Pacini, E. Nectary structure, nectar secretion patterns and nectar composition in two Helleborus species. Plant Biol. 1999, 1, 560–568. [Google Scholar] [CrossRef]
- Bernardello, G.; Galetto, L.; Anderson, G.J. Floral nectary structure and nectar chemical composition of some species from Robinson Crusoe Island (Chile). Can. J. Bot. 2000, 78, 862–872. [Google Scholar] [CrossRef]
- Konarska, A. Comparison of the structure of floral nectaries in two Euonymus L. species (Celastraceae). Protoplasma 2015, 252, 901–910. [Google Scholar] [CrossRef]
- Radhika, V.; Kost, C.; Boland, W.; Heil, M. The Role of Jasmonates in Floral Nectar Secretion. PLoS ONE 2010, 5, e9265. [Google Scholar] [CrossRef] [PubMed]
- Fahn, A. Structure and function of secretory cells. In Advances in Botanical Research; Academic Press: New York, NY, SUA, 2000; Volume 31, pp. 37–75. [Google Scholar]
- Pacini, E.; Nepi, M.; Vesprini, J.L. Nectar biodiversity: A short review. Plant Syst. Evol. 2003, 238, 7–21. [Google Scholar] [CrossRef]
- Erbar, C. Nectar secretion and nectaries in basal angiosperms, magnoliids and non-core eudicots and a comparison with core eudicots. Plant Divers. Evol. 2014, 131, 63–143. [Google Scholar] [CrossRef]
- Sandvik, S.M.; Totland, O. Quantitative importance of staminodes for female reproductive success in Parnassia palustris under contrasting environmental conditions. Can. J. Bot. 2003, 81, 49–56. [Google Scholar] [CrossRef]
- Ollerton, J.; Winfree, R.; Tarrant, S. How many flowering plants are pollinated by animals? Oikos 2011, 120, 321–326. [Google Scholar] [CrossRef]
- Kram, B.W.; Xu, W.W.; Carter, C.J. Uncovering the Arabidopsis thaliana nectary transcriptome: Investigation of differential gene expression in floral nectariferous tissues. BMC Plant Biol. 2009, 9, 92. [Google Scholar] [CrossRef]
- Burquez, A.; Corbet, S.A. Do Flowers Reabsorb Nectar? Funct. Ecol. 1991, 5, 369–379. [Google Scholar] [CrossRef]
- Fahn, A. Ultrastructure of Nectaries in Relation to Nectar Secretion. Am. J. Bot. 1979, 66, 977–985. [Google Scholar] [CrossRef]
- Peng, Y.B.; Li, Y.Q.; Hao, Y.J.; Xu, Z.H.; Bai, S.N. Nectar production and transportation in the nectaries of the female Cucumis sativus L. flower during anthesis. Protoplasma 2004, 224, 71–78. [Google Scholar] [CrossRef]
- Razem, F.A.; Davis, A.R. Anatomical and ultrastructural changes of the floral nectary of Pisum sativum L. during flower development. Protoplasma 1999, 206, 57–72. [Google Scholar] [CrossRef]
- Chen, L.Q. SWEET sugar transporters for phloem transport and pathogen nutrition. New Phytol. 2014, 201, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Q.; Qu, X.Q.; Hou, B.H.; Sosso, D.; Osorio, S.; Fernie, A.R.; Frommer, W.B. Sucrose Efflux Mediated by SWEET Proteins as a Key Step for Phloem Transport. Science 2012, 335, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Q.; Cheung, L.S.; Feng, L.; Tanner, W.; Frommer, W.B. Transport of Sugars. Annu. Rev. Biochem. 2015, 84, 865–894. [Google Scholar] [CrossRef]
- Vassilyev, A.E. On the mechanisms of nectar secretion: Revisited. Ann. Bot. 2010, 105, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, E.; Tunes, P.; de Almeida, L.D.; Di Stasi, L.C.; Dotterl, S.; Machado, S.R. Nectar Replaced by Volatile Secretion: A Potential New Role for Nectarless Flowers in a Bee-Pollinated Plant Species. Front. Plant Sci. 2018, 9, 1243. [Google Scholar] [CrossRef]
- Konarska, A.; Masierowska, M. Structure of floral nectaries and female-biased nectar production in protandrous species Geranium macrorrhizum and Geranium phaeum. Protoplasma 2020, 257, 501–523. [Google Scholar] [CrossRef]
- Nepi, M.; Stpiczynska, M. Nectar resorption and translocation in Cucurbita pepo L. and Platanthera chlorantha Custer (Rchb.). Plant Biol. 2007, 9, 93–100. [Google Scholar] [CrossRef]
- Ruhlmann, J.M.; Kram, B.W.; Carter, C.J. CELL WALL INVERTASE 4 is required for nectar production in Arabidopsis. J. Exp. Bot. 2010, 61, 395–404. [Google Scholar] [CrossRef]
- Chong, J.; Piron, M.C.; Meyer, S.; Merdinoglu, D.; Bertsch, C.; Mestre, P. The SWEET family of sugar transporters in grapevine: VvSWEET4 is involved in the interaction with Botrytis cinerea. J. Exp. Bot. 2014, 65, 6589–6601. [Google Scholar] [CrossRef]
- Feng, C.Y.; Han, J.X.; Han, X.X.; Jiang, J. Genome-wide identification, phylogeny, and expression analysis of the SWEET gene family in tomato. Gene 2015, 573, 261–272. [Google Scholar] [CrossRef]
- Patil, G.; Valliyodan, B.; Deshmukh, R.; Prince, S.; Nicander, B.; Zhao, M.Z.; Sonah, H.; Song, L.; Lin, L.; Chaudhary, J.; et al. Soybean (Glycine max) SWEET gene family: Insights through comparative genomics, transcriptome profiling and whole genome re-sequence analysis. Bmc Genom. 2015, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Wang, S.P. Rice MtN3/Saliva/SWEET Family Genes and Their Homologs in Cellular Organisms. Mol. Plant 2013, 6, 665–674. [Google Scholar] [CrossRef]
- Gautam, T.; Saripalli, G.; Gahlaut, V.; Kumar, A.; Sharma, P.K.; Balyan, H.S.; Gupta, P.K. Further studies on sugar transporter (SWEET) genes in wheat (Triticum aestivum L.). Mol. Biol. Rep. 2019, 46, 2327–2353. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Zhang, A.H.; Jiang, J. Gene expression patterns of invertase gene families and modulation of the inhibitor gene in tomato sucrose metabolism. Genet. Mol. Res. 2013, 12, 3412–3420. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.K.; Yang, L.; Zhu, H.J.; Zhou, J.; Sun, H.; Gong, H.J. Structure and Expression Analysis of Sucrose Phosphate Synthase, Sucrose Synthase and Invertase Gene Families in Solanum lycopersicum. Int. J. Mol. Sci. 2021, 22, 4698. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, J.; Štěpánek, J. Clonality as a part of the evolution process inTaraxacum. Folia Geobot. 1994, 29, 265–275. [Google Scholar] [CrossRef]
- Kirschner, J.; Stepanek, J. New sections in Taraxacum. Folia Geobot. 2004, 39, 259–274. [Google Scholar] [CrossRef]
- Uhlemann, I.; Kirschner, J.; Stepanek, J. The genus Taraxacum (Asteraceae) in the southern hemisphere. I. The section Antarctica Handel-Mazzetti and notes on dandelions of Australasia. Folia Geobot. 2004, 39, 205–220. [Google Scholar] [CrossRef]
- Vasut, R.J.; Stepanek, J.; Kirschner, J. Two new apomictic Taraxacum microspecies of the section Erythrosperma from central Europe. Preslia 2005, 77, 197–210. [Google Scholar]
- Richards, A.J. Genetic variability in obligate apomicts of the genusTaraxacum. Folia Geobot. 1996, 31, 405–414. [Google Scholar] [CrossRef]
- RICHARDS, A.J. The origin of Taraxacum agamospecies. Bot. J. Linn. Soc. 2008, 66, 189–211. [Google Scholar] [CrossRef]
- Barrett, S.C.H.; van Dijk, P.J.; Vinkenoog, R. Ecological and evolutionary opportunities of apomixis: Insights from Taraxacum and Chondrilla. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 1120–1121. [Google Scholar]
- Çelemli, Ö.G.; Özkök, A.; Özenirler, Ç.; Mayda, N.; Sorkun, K. Dandelion honey: A new monofloral honey record for Turkey. 2018, 87–93. Uludağ Arıcılık Dergisi 2018, 18, 87–93. [Google Scholar] [CrossRef]
- Margaoan, R.; Topal, E.; Balkanska, R.; Yucel, B.; Oravecz, T.; Cornea-Cipcigan, M.; Vodnar, D.C. Monofloral Honeys as a Potential Source of Natural Antioxidants, Minerals and Medicine. Antioxidants 2021, 10, 1023. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.M.; Ouvrard, P.; Baldock, K.C.; Baude, M.; Goddard, M.A.; Kunin, W.E.; Mitschunas, N.; Memmott, J.; Morse, H.; Nikolitsi, M.; et al. Food for Pollinators: Quantifying the Nectar and Pollen Resources of Urban Flower Meadows. PLoS ONE 2016, 11, e0158117. [Google Scholar] [CrossRef]
- Szabo, T.I. Nectar Secretion in Dandelion. J. Apic. Res. 1984, 23, 204–208. [Google Scholar] [CrossRef]
- Pacini, E.; Nepi, M. Nectar Production and Presentation. In Nectaries and Nectar; Springer: Dordrecht, The Netherlands, 2007. [Google Scholar]
- Carlson, J.E.; Harms, K. E The evolution of gender-biased nectar production in hermaphroditic plants. Bot. Rev. 2006, 72, 179–205. [Google Scholar] [CrossRef]
- Bernardello, G. A Systematic Survey of Floral Nectaries. In Nectaries and Nectar; Springer: Dordrecht, The Netherlands, 2007. [Google Scholar]
- Shibaike, H.; Akiyama, H.; Uchiyama, S.; Kasai, K.; Morita, T. Hybridization between European and Asian dandelions (Taraxacum section Ruderalia and section Mongolica) 2. Natural hybrids in Japan detected by chloroplast DNA marker. J. Plant Res. 2002, 115, 321–328. [Google Scholar] [CrossRef]
- Tas, I.C.Q.; Van Dijk, P.J. Crosses between sexual and apomictic dandelions (Taraxacum). I. The inheritance of apomixis. Heredity 1999, 83, 707–714. [Google Scholar] [CrossRef]
- Wind, J.; Smeekens, S.; Hanson, J. Sucrose: Metabolite and signaling molecule. Phytochemistry 2010, 71, 1610–1614. [Google Scholar] [CrossRef]
- Ayre, B.G. Membrane-Transport Systems for Sucrose in Relation to Whole-Plant Carbon Partitioning. Mol. Plant 2011, 4, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.M. SWEET! The Pathway Is Complete. Science 2012, 335, 173–174. [Google Scholar] [CrossRef]
- Chen, L.Q.; Hou, B.H.; Lalonde, S.; Takanaga, H.; Hartung, M.L.; Qu, X.Q.; Guo, W.J.; Kim, J.G.; Underwood, W.; Chaudhuri, B.; et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 2010, 468, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Eom, J.S.; Chen, L.Q.; Sosso, D.; Julius, B.T.; Lin, I.W.; Qu, X.Q.; Braun, D.M.; Frommer, W.B. SWEETs, transporters for intracellular and intercellular sugar translocation. Curr. Opin. Plant Biol. 2015, 25, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Roldan-Serrano, A.S.; Guerra-Sanz, J.M. Reward attractions of zucchini flowers (Cucurbita pepo L.) to bumblebees (Bombus terrestris L.). Eur. J. Hortic. Sci. 2005, 70, 23–28. [Google Scholar] [CrossRef]
- Lin, Q.; Zhong, Q.; Zhang, Z. Identification and functional analysis of SWEET gene family in Averrhoa carambola L. fruits during ripening. PeerJ 2021, 9, e11404. [Google Scholar] [CrossRef]
- Yao, T.S.; Xie, R.J.; Zhou, Y.; Hu, J.H.; Gao, Y.; Zhou, C.Y. Genome-Wide Identification of SWEET Gene Family and Its Response to Abiotic Stresses in Valencia Sweet Orange. Plant Mol. Biol. Rep. 2021, 39, 546–556. [Google Scholar] [CrossRef]
- Huang, D.; Chen, Y.; Qin, Q. Genome-wide identification and expression analysis of SWEET gene family in daylily (Hemerocallis fulva) and functional analysis of HfSWEET17 in response to cold stress. BMC Plant Biol. 2022, 22, 211. [Google Scholar] [CrossRef]
- Yin, Q.; Zhu, L.; Du, P.; Fan, C.; Wang, J.; Zhang, B.; Li, H. Comprehensive analysis of SWEET family genes in Eucalyptus (Eucalyptus grandis). Biotechnol. Biotechnol. Equip. 2020, 34, 595–604. [Google Scholar] [CrossRef]
- Jiang, L.; Song, C.; Zhu, X.; Yang, J.K. SWEET Transporters and the Potential Functions of These Sequences in Tea (Camellia sinensis). Front. Genet. 2021, 12, 655843. [Google Scholar] [CrossRef]
- Zhang, R.; Niu, K.J.; Ma, H.L. Identification and Expression Analysis of the SWEET Gene Family from Poa pratensis Under Abiotic Stresses. DNA Cell Biol. 2020, 39, 1606–1620. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.T.; Lyu, W.Y.; Tian, S.H.; Zou, X.H.; Zhang, L.Q.; Gao, Q.H.; Ni, D.A.; Duan, K. The SWEET family genes in strawberry: Identification and expression profiling during fruit development. S. Afr. J. Bot. 2019, 125, 176–187. [Google Scholar] [CrossRef]
- Xuan, C.Q.; Lan, G.P.; Si, F.F.; Zeng, Z.L.; Wang, C.X.; Yadav, V.; Wei, C.H.; Zhang, X. Systematic Genome-Wide Study and Expression Analysis of SWEET Gene Family: Sugar Transporter Family Contributes to Biotic and Abiotic Stimuli in Watermelon. Int. J. Mol. Sci. 2021, 22, 8407. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.H.; Wang, D.; Qin, Y.Q.; Ma, A.N.; Fu, J.X.; Qin, Y.H.; Hu, G.B.; Zhao, J.T. Genome-wide identification and expression analysis of SWEET gene family in Litchi chinensis reveal the involvement of LcSWEET2a/3b in early seed development. BMC Plant Biol. 2019, 19, 1–13. [Google Scholar] [CrossRef]
- Qin, N.N.; Gao, Y.; Cheng, X.J.; Yang, Y.; Wu, J.; Wang, J.Y.; Li, S.; Xing, G.M. Genome-wide identification of CLE gene family and their potential roles in bolting and fruit bearing in cucumber (Cucumis sativus L.). BMC Plant Biol. 2021, 21, 1–18. [Google Scholar] [CrossRef]
- Hu, L.P.; Zhang, F.; Song, S.H.; Tang, X.W.; Xu, H.; Liu, G.M.; Wang, Y.G.; He, H.J. Genome-wide identification, characterization, and expression analysis of the SWEET gene family in cucumber. J. Integr. Agric. 2017, 16, 1486–1501. [Google Scholar] [CrossRef]
- Zhao, L.J.; Yao, J.B.; Chen, W.; Li, Y.; Lo, Y.J.; Guo, Y.; Wang, J.Y.; Yuan, L.; Liu, Z.Y.; Zhang, Y.S. A genome-wide analysis of SWEET gene family in cotton and their expressions under different stresses. J. Cotton Res. 2018, 1, 1–15. [Google Scholar] [CrossRef]
- Hu, B.; Wu, H.; Huang, W.F.; Song, J.B.; Zhou, Y.; Lin, Y.J. SWEET Gene Family in Medicago truncatula: Genome-Wide Identification, Expression and Substrate Specificity Analysis. Plants 2019, 8, 338. [Google Scholar] [CrossRef]
- Sui, J.L.; Xiao, X.H.; Qi, J.Y.; Fang, Y.J.; Tang, C.R. The SWEET gene family in Hevea brasiliensis—Its evolution and expression compared with four other plant species. Febs Open Bio 2017, 7, 1943–1959. [Google Scholar] [CrossRef]
- Jian, H.J.; Lu, K.; Yang, B.; Wang, T.Y.; Zhang, L.; Zhang, A.X.; Wang, J.; Liu, L.Z.; Qu, C.M.; Li, J.N. Genome-Wide Analysis and Expression Profiling of the SUC and SWEET Gene Families of Sucrose Transporters in Oilseed Rape (Brassica napus L.). Front. Plant Sci. 2016, 7, 1464. [Google Scholar] [CrossRef]
- Wei, X.; Liu, F.; Chen, C.; Ma, F.; Li, M. The Malus domestica sugar transporter gene family: Identifications based on genome and expression profiling related to the accumulation of fruit sugars. Front. Plant Sci. 2014, 5, 569. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Sun, P.; Liu, Q.; Miao, Y.; Liu, J.; Zhang, K.; Hu, W.; Zhang, J.; Wang, J.; Wang, Z.; et al. Genome-wide analyses of SWEET family proteins reveal involvement in fruit development and abiotic/biotic stress responses in banana. Sci. Rep. 2017, 7, 3536. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, Y.; Yang, X.; Xing, J.; Qi, J.; Zhang, S.; Zhang, Y.; Ye, D.; Tang, C. Genome-wide analysis of the SWEET genes in Taraxacum kok-saghyz Rodin: An insight into two latex-abundant isoforms. Plant Physiol. Bioch 2023, 194, 440–448. [Google Scholar] [CrossRef]
- Lin, I.W.; Sosso, D.; Chen, L.Q.; Gase, K.; Kim, S.G.; Kessler, D.; Klinkenberg, P.M.; Gorder, M.K.; Hou, B.H.; Qu, X.Q.; et al. Nectar secretion requires sucrose phosphate synthases and the sugar transporter SWEET9. Nature 2014, 508, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Sosso, D.; Luo, D.P.; Li, Q.B.; Sasse, J.; Yang, J.L.; Gendrot, G.; Suzuki, M.; Koch, K.E.; McCarty, D.R.; Chourey, P.S.; et al. Seed filling in domesticated maize and rice depends on SWEET-mediated hexose transport. Nat. Genet. 2015, 47, 1489–1493. [Google Scholar] [CrossRef]
- Guo, C.Y.; Li, H.Y.; Xia, X.Y.; Liu, X.Y.; Yang, L. Functional and evolution characterization of SWEET sugar transporters in Ananas comosus. Biochem. Biophys. Res. Commun. 2018, 496, 407–414. [Google Scholar] [CrossRef]
- Kanno, Y.; Oikawa, T.; Chiba, Y.; Ishimaru, Y.; Shimizu, T.; Sano, N.; Koshiba, T.; Kamiya, Y.; Ueda, M.; Seo, M. AtSWEET13 and AtSWEET14 regulate gibberellin-mediated physiological processes. Nat. Commun. 2016, 7, 13245. [Google Scholar] [CrossRef] [PubMed]
- Andres, F.; Kinoshita, A.; Kalluri, N.; Fernandez, V.; Falavigna, V.S.; Cruz, T.M.D.; Jang, S.; Chiba, Y.; Seo, M.; Mettler-Altmann, T.; et al. The sugar transporter SWEET10 acts downstream of FLOWERING LOCUS T during floral transition of Arabidopsis thaliana. Bmc Plant Biol. 2020, 20, 1–14. [Google Scholar] [CrossRef]
- Abelenda, J.A.; Bergonzi, S.; Oortwijn, M.; Sonnewald, S.; Du, M.R.; Visser, R.G.F.; Sonnewald, U.; Bachem, C.W.B. Source-Sink Regulation Is Mediated by Interaction of an FT Homolog with a SWEET Protein in Potato. Curr. Biol. 2019, 29, 1178–1186.e6. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, D.C.; Miao, Q.S.; Yang, J.; Xuan, Y.H.; Hu, Y.B. Essential Role of Sugar Transporter OsSWEET11 During the Early Stage of Rice Grain Filling. Plant Cell Physiol. 2017, 58, 863–873. [Google Scholar] [CrossRef]
- Yang, J.L.; Luo, D.P.; Yang, B.; Frommer, W.B.; Eom, J.S. SWEET11 and 15 as key players in seed filling in rice. New Phytol. 2018, 218, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Melkus, G.; Rolletschek, H.; Fuchs, J.; Radchuk, V.; Grafahrend-Belau, E.; Sreenivasulu, N.; Rutten, T.; Weier, D.; Heinzel, N.; Schreiber, F.; et al. Dynamic C/H NMR imaging uncovers sugar allocation in the living seed. Plant Biotechnol. J. 2011, 9, 1022–1037. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.H.; Endo, A.; Zhou, L.; Penney, J.; Chen, H.C.; Arroyo, A.; Leon, P.; Nambara, E.; Asami, T.; Seo, M.; et al. A unique short-chain dehydrogenase/reductase in Arabidopsis glucose signaling and abscisic acid biosynthesis and functions. Plant Cell 2002, 14, 2723–2743. [Google Scholar] [CrossRef] [PubMed]
- Klemens, P.A.W.; Patzke, K.; Deitmer, J.; Spinner, L.; Le Hir, R.; Bellini, C.; Bedu, M.; Chardon, F.; Krapp, A.; Neuhaus, H.E. Overexpression of the Vacuolar Sugar Carrier AtSWEET16 Modifies Germination, Growth, and Stress Tolerance in Arabidopsis. Plant Physiol. 2013, 163, 1338–1352. [Google Scholar] [CrossRef]
- Klemens, P.A.W.; Patzke, K.; Krapp, A.; Chardon, F.; Neuhaus, H.E. SWEET16 and SWEET17, two novel vacuolar sugar carriers with impact on cellular sugar homeostasis and plant traits. Biochem. Cell Biol. 2014, 92, 589. [Google Scholar]
- Wu, Y.T.; Wu, P.Z.; Xu, S.M.; Chen, Y.P.; Li, M.R.; Wu, G.J.; Jiang, H.W. Genome-Wide Identification, Expression Patterns and Sugar Transport of the Physic Nut Gene Family and a Functional Analysis of in Arabidopsis. Int. J. Mol. Sci. 2022, 23, 5391. [Google Scholar] [CrossRef]
- Solhaug, E.M.; Roy, R.; Chatt, E.C.; Klinkenberg, P.M.; Mohd-Fadzil, N.A.; Hampton, M.; Nikolau, B.J.; Carter, C.J. An integrated transcriptomics and metabolomics analysis of the Cucurbita pepo nectary implicates key modules of primary metabolism involved in nectar synthesis and secretion. Plant Direct 2019, 3, e00120. [Google Scholar] [CrossRef]
- Nishanth, M.J.; Sheshadri, S.A.; Rathore, S.S.; Srinidhi, S.; Simon, B. Expression analysis of Cell wall invertase under abiotic stress conditions influencing specialized metabolism in Catharanthus roseus. Sci. Rep. 2018, 8, 15059. [Google Scholar] [CrossRef]
- Poschet, G.; Hannich, B.; Raab, S.; Jungkunz, I.; Klemens, P.A.W.; Krueger, S.; Wic, S.; Neuhaus, H.E.; Buttner, M. A Novel Arabidopsis Vacuolar Glucose Exporter Is Involved in Cellular Sugar Homeostasis and Affects the Composition of Seed Storage Compounds. Plant Physiol. 2011, 157, 1664–1676. [Google Scholar] [CrossRef]
- Cho, J.I.; Lee, S.K.; Ko, S.; Kim, H.K.; Jun, S.H.; Lee, Y.H.; Bhoo, S.H.; Lee, K.W.; An, G.; Hahn, T.R.; et al. Molecular cloning and expression analysis of the cell-wall invertase gene family in rice (Oryza sativa L.). Plant Cell Rep. 2005, 24, 225–236. [Google Scholar] [CrossRef]
- Chen, Z.; Gao, K.; Su, X.; Rao, P.; An, X. Genome-Wide Identification of the Invertase Gene Family in Populus. PLoS ONE 2015, 10, e0138540. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Geng, M.T.; Wu, X.H.; Liu, J.; Li, R.M.; Hu, X.W.; Guo, J.C. Genome-Wide Identification, 3D Modeling, Expression and Enzymatic Activity Analysis of Cell Wall Invertase Gene Family from Cassava (Manihot esculenta Crantz). Int. J. Mol. Sci. 2014, 15, 7313–7331. [Google Scholar] [CrossRef]
- Li, J.; Foster, R.; Ma, S.; Liao, S.J.; Bliss, S.; Kartika, D.; Wang, L.; Wu, L.; Eamens, A.L.; Ruan, Y.L. Identification of transcription factors controlling cell wall invertase gene expression for reproductive development via bioinformatic and transgenic analyses. Plant J. 2021, 106, 1058–1074. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Xu, X.; Zhang, L.; Zhang, H.; Lin, L.; Wang, Q.; Li, Q.; Ge, S.; Lu, B.R.; Wang, W.; et al. Duplication and independent selection of cell-wall invertase genes GIF1 and OsCIN1 during rice evolution and domestication. BMC Evol. Biol. 2010, 10, 108. [Google Scholar] [CrossRef]
- Su, A.G.; Song, W.; Shi, Z.; Zhao, Y.X.; Xing, J.F.; Zhang, R.Y.; Li, C.H.; Luo, M.J.; Wang, J.D.; Zhao, J.R. Exploring differentially expressed genes associated with fertility instability of S-type cytoplasmic male-sterility in maize by RNA-seq. J. Integr. Agric. 2017, 16, 1689–1699. [Google Scholar] [CrossRef]
- Edstam, M.M.; Edqvist, J. Involvement of GPI-anchored lipid transfer proteins in the development of seed coats and pollen in Arabidopsis thaliana. Physiol. Plant. 2014, 152, 32–42. [Google Scholar] [CrossRef]
- Engelke, T.; Hirsche, J.; Roitsch, T. Anther-specific carbohydrate supply and restoration of metabolically engineered male sterility. J. Exp. Bot. 2010, 61, 2693–2706. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.L.; Lindhout, P. Domestication and breeding of tomatoes: What have we gained and what can we gain in the future? Ann. Bot. 2007, 100, 1085–1094. [Google Scholar] [CrossRef]
- Santos, E.A.; Souza, M.M.; Abreu, P.P.; da Conceicao, L.D.H.C.S.; Araujo, I.S.; Viana, A.P.; de Almeida, A.A.F.; Freitas, J.C.D. Confirmation and characterization of interspecific hybrids of Passiflora L. (Passifloraceae) for ornamental use. Euphytica 2012, 184, 389–399. [Google Scholar] [CrossRef]
- Khan, M.M.R.; Isshiki, S. Cytoplasmic Male Sterility in Eggplant. Hortic. J. 2016, 85, 1–7. [Google Scholar] [CrossRef]
- Ma, H. Molecular genetic analyses of microsporogenesis and microgametogenesis in flowering plants. Annu. Rev. Plant Biol. 2005, 56, 393–434. [Google Scholar] [CrossRef] [PubMed]
- Breitler, J.C.; Campa, C.; Georget, F.; Bertrand, B.; Etienne, H. A single-step method for RNA isolation from tropical crops in the field. Sci. Rep. 2016, 6, 38368. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Davidson, N.M.; Hawkins, A.D.K.; Oshlack, A. SuperTranscripts: A data driven reference for analysis and visualisation of transcriptomes. Genome Biol. 2017, 18, 148. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simao, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Poole, R.L. The TAIR database. Methods Mol. Biol. 2007, 406, 179–212. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST plus: Architecture and applications. Bmc Bioinform. 2009, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 1–19. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Chen, Y.; Lun, A.T.; Smyth, G.K. From reads to genes to pathways: Differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Research 2016, 5, 1438. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; Garcia-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef]
- Tukey, J.W. Comparing Individual Means in the Analysis of Variance. Biometrics 1949, 5, 99–114. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.no | Species | Longest Transcripts | N50 | Unigenes | Cd-Hit-Est Unigenes | TAIR | Swiss-Prot | KEGG |
|---|---|---|---|---|---|---|---|---|
| 1 | T. ohwianum | 122,494 | 1635 | 64,296 | 27,691 | 16,431 | 47,659 | 35,261 |
| 2 | T. officinale | 120,911 | 938.75 | 58,924 | 29,511 | 15,782 | 35,788 | 35,165 |
| 3 | T. mongolicum | 129,433 | 1053.75 | 69,234 | 32,706 | 22,900 | 48,092 | 39,386 |
| 4 | T. coreanum | 156,377 | 929.84 | 73,698 | 31,318 | 24,678 | 69,375 | 44,722 |
| 5 | T. hallaisanense | 96,360 | 927.65 | 59,599 | 29,039 | 17,897 | 33,334 | 28,375 |
| 6 | combined | 397,344 | 1304 | 197,473 | 58,339 | 53,564 | 65,149 | 58,783 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claude, S.-J.; Park, S.; Park, S.-J.; Park, S. Unraveling the Nectar Secretion Pathway and Floral-Specific Expression of SWEET and CWIV Genes in Five Dandelion Species Through RNA Sequencing. Plants 2025, 14, 1718. https://doi.org/10.3390/plants14111718
Claude S-J, Park S, Park S-J, Park S. Unraveling the Nectar Secretion Pathway and Floral-Specific Expression of SWEET and CWIV Genes in Five Dandelion Species Through RNA Sequencing. Plants. 2025; 14(11):1718. https://doi.org/10.3390/plants14111718
Chicago/Turabian StyleClaude, Sivagami-Jean, Sunmi Park, Seong-Jun Park, and SeonJoo Park. 2025. "Unraveling the Nectar Secretion Pathway and Floral-Specific Expression of SWEET and CWIV Genes in Five Dandelion Species Through RNA Sequencing" Plants 14, no. 11: 1718. https://doi.org/10.3390/plants14111718
APA StyleClaude, S.-J., Park, S., Park, S.-J., & Park, S. (2025). Unraveling the Nectar Secretion Pathway and Floral-Specific Expression of SWEET and CWIV Genes in Five Dandelion Species Through RNA Sequencing. Plants, 14(11), 1718. https://doi.org/10.3390/plants14111718