
Integrative Physiological and Transcriptomic Analysis Reveals Metabolic Adaptation and Cold-Tolerance Marker Development in Winter Rye Under Low-Temperature Stress


Abstract
1. Introduction
2. Results
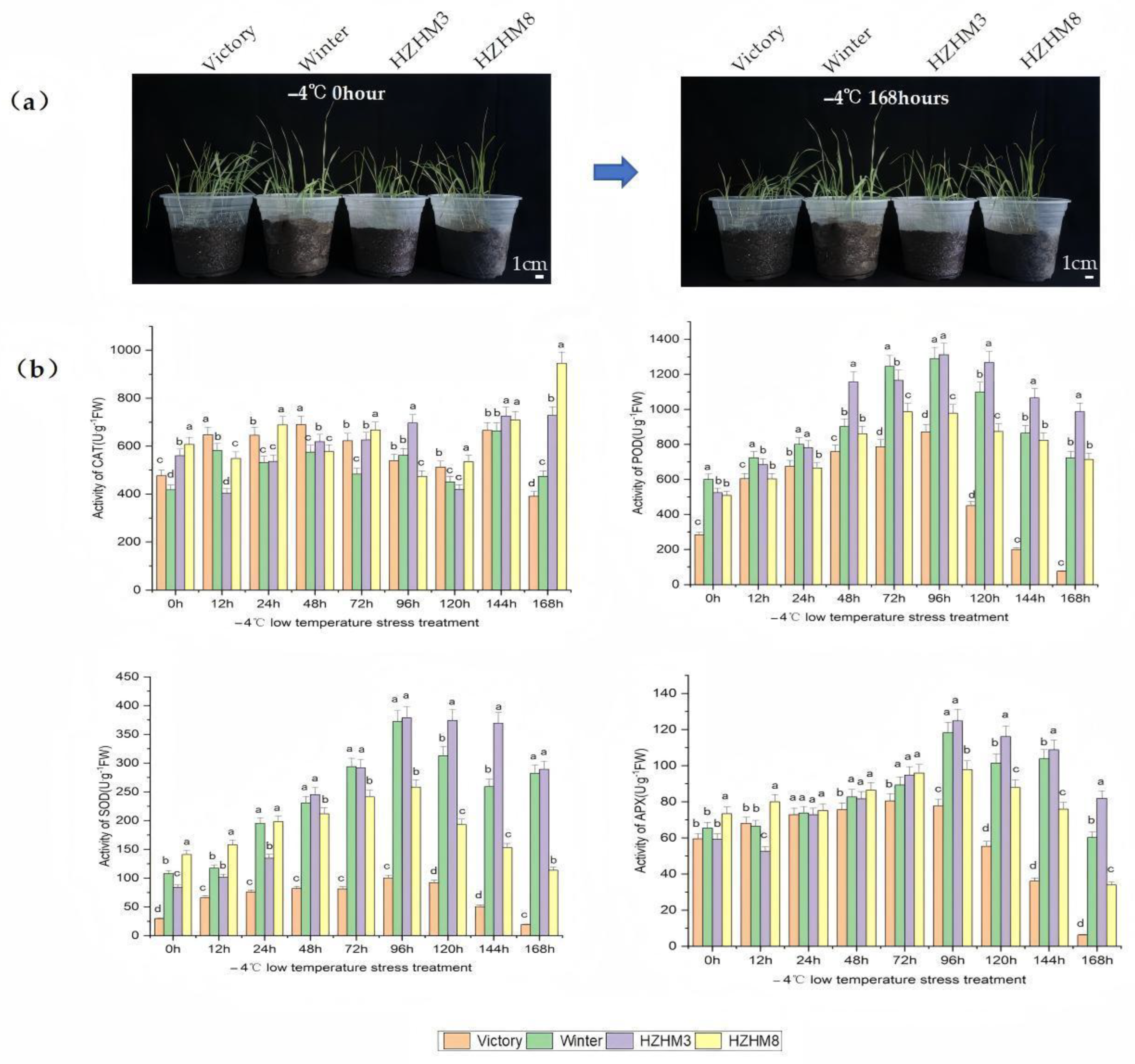
2.1. Physiological Responses and Cold Tolerance Evaluation of Different Rye Varieties Under Low-Temperature Stress
2.2. Transcriptome Analysis and Differentially Expressed Gene Screening in “Winter” Rye Under −4 °C Low-Temperature Stress
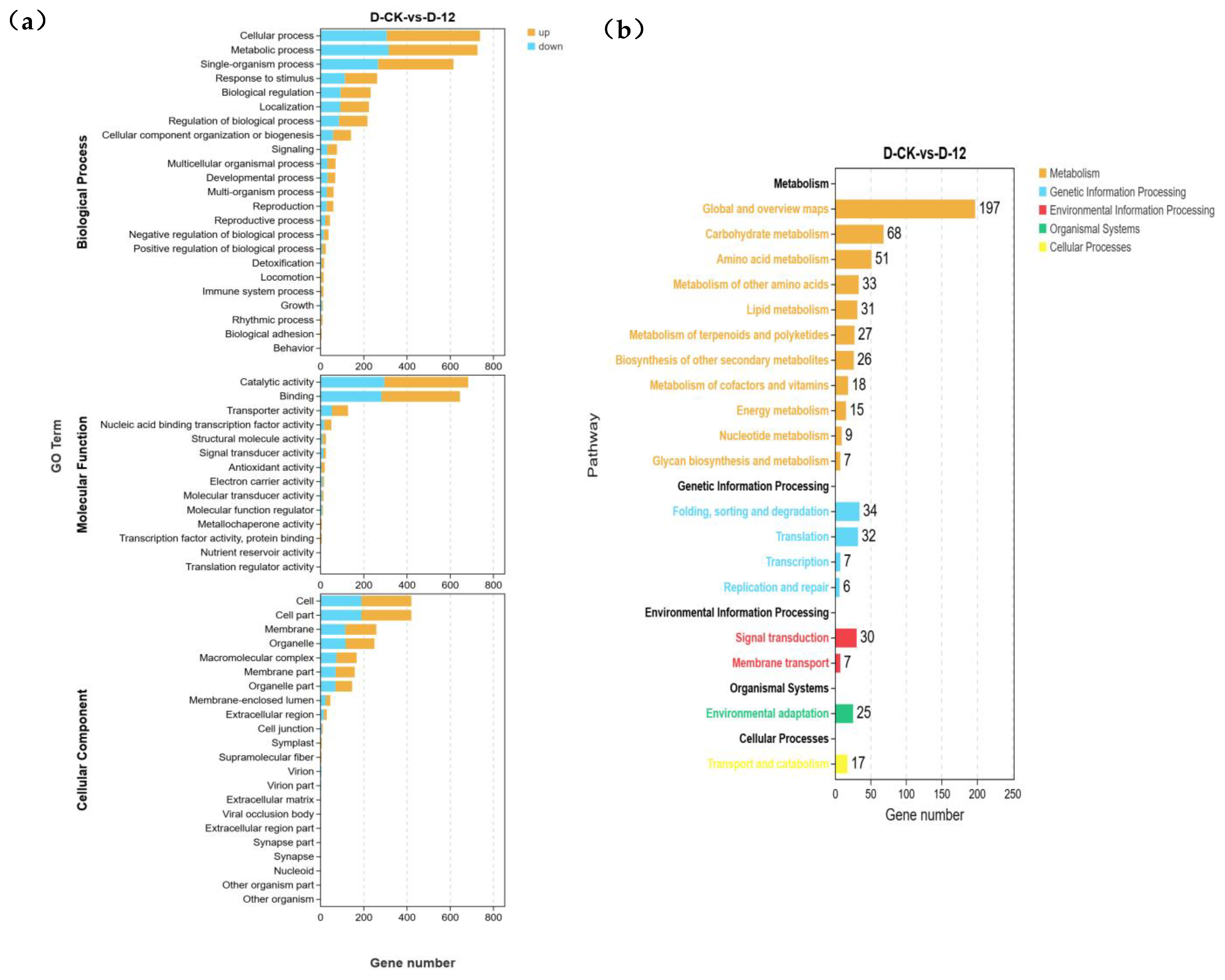
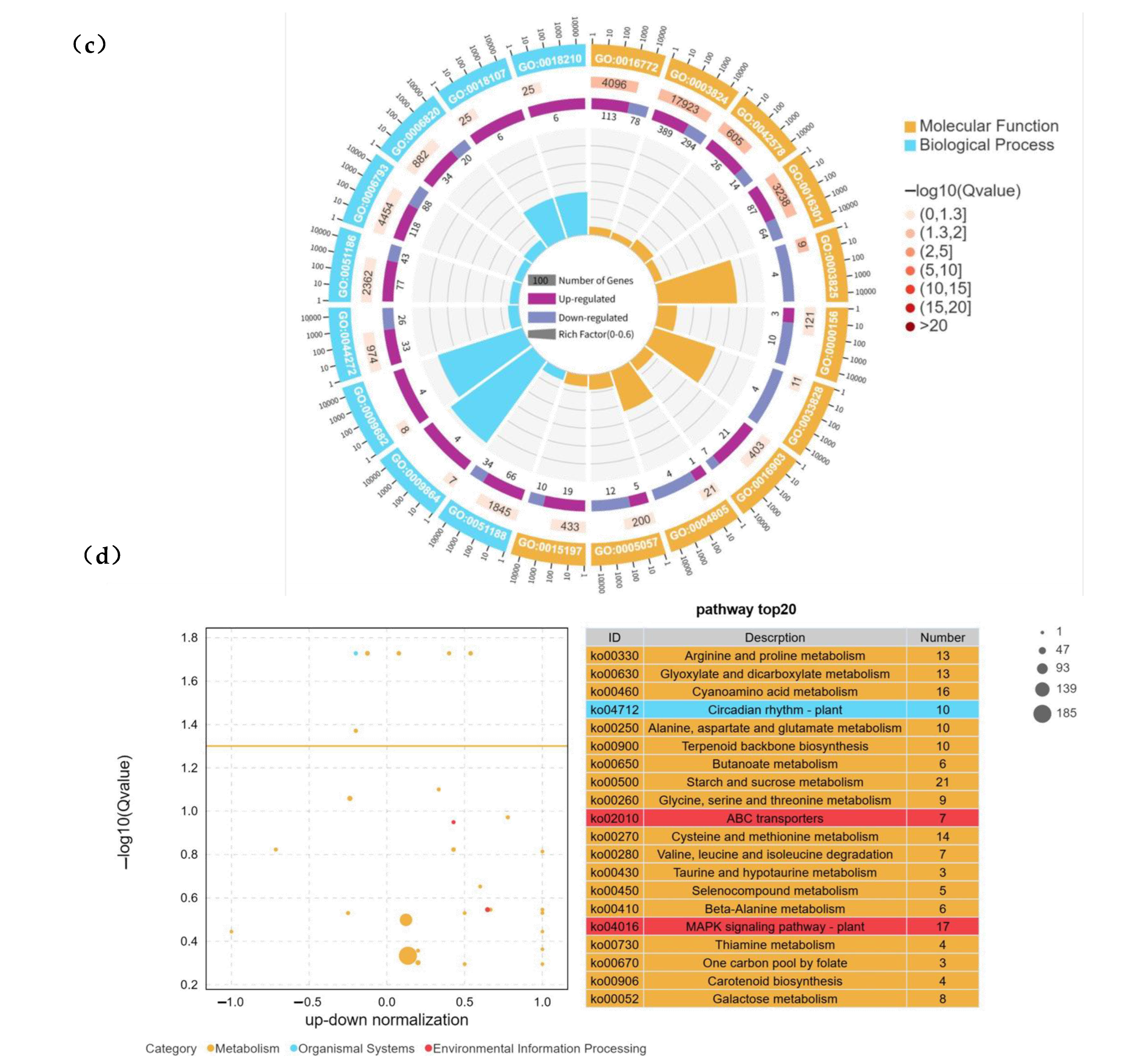
2.3. GO and KEGG Enrichment Analysis of Differentially Expressed Genes in “Winter” Rye Under Low-Temperature Stress
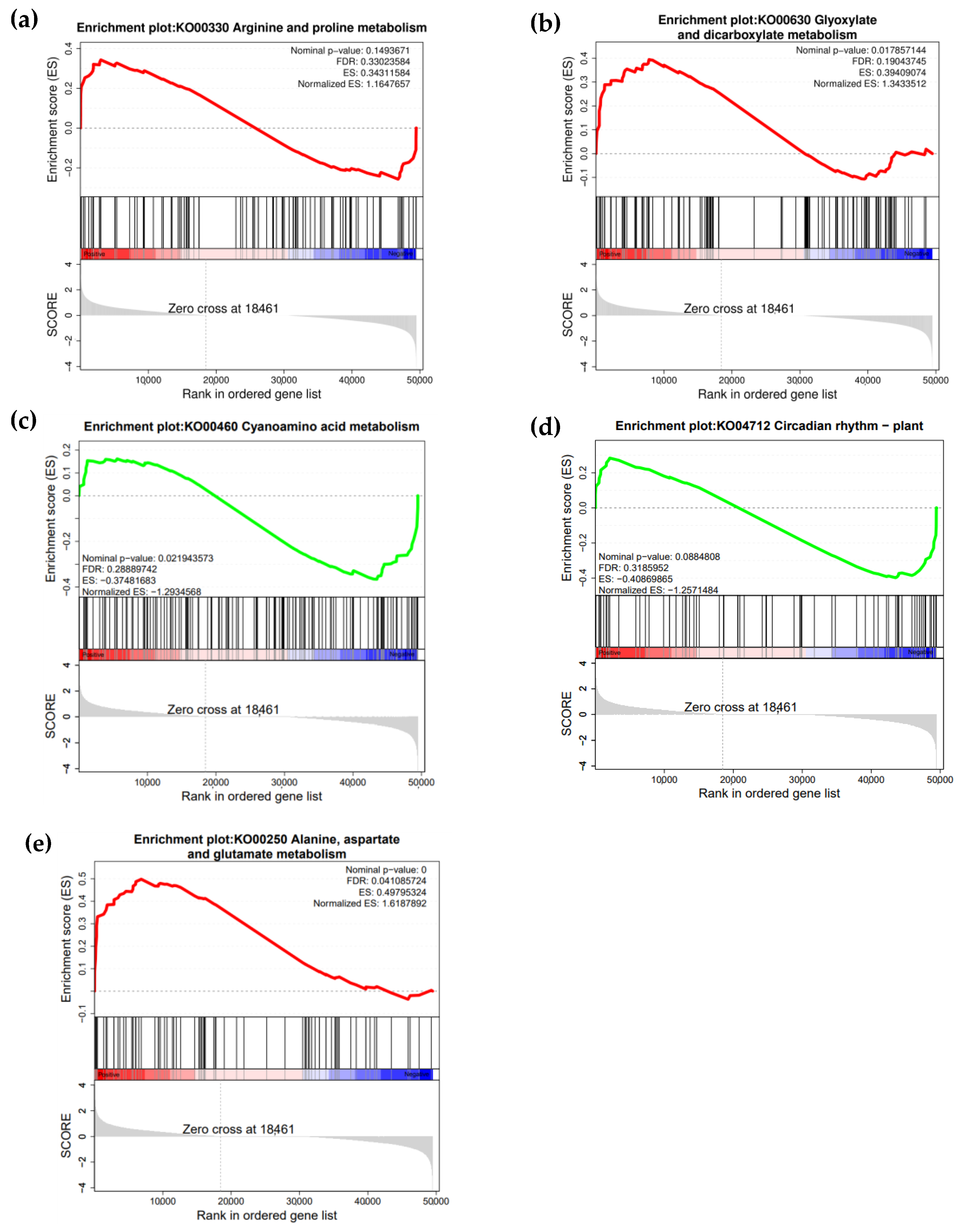
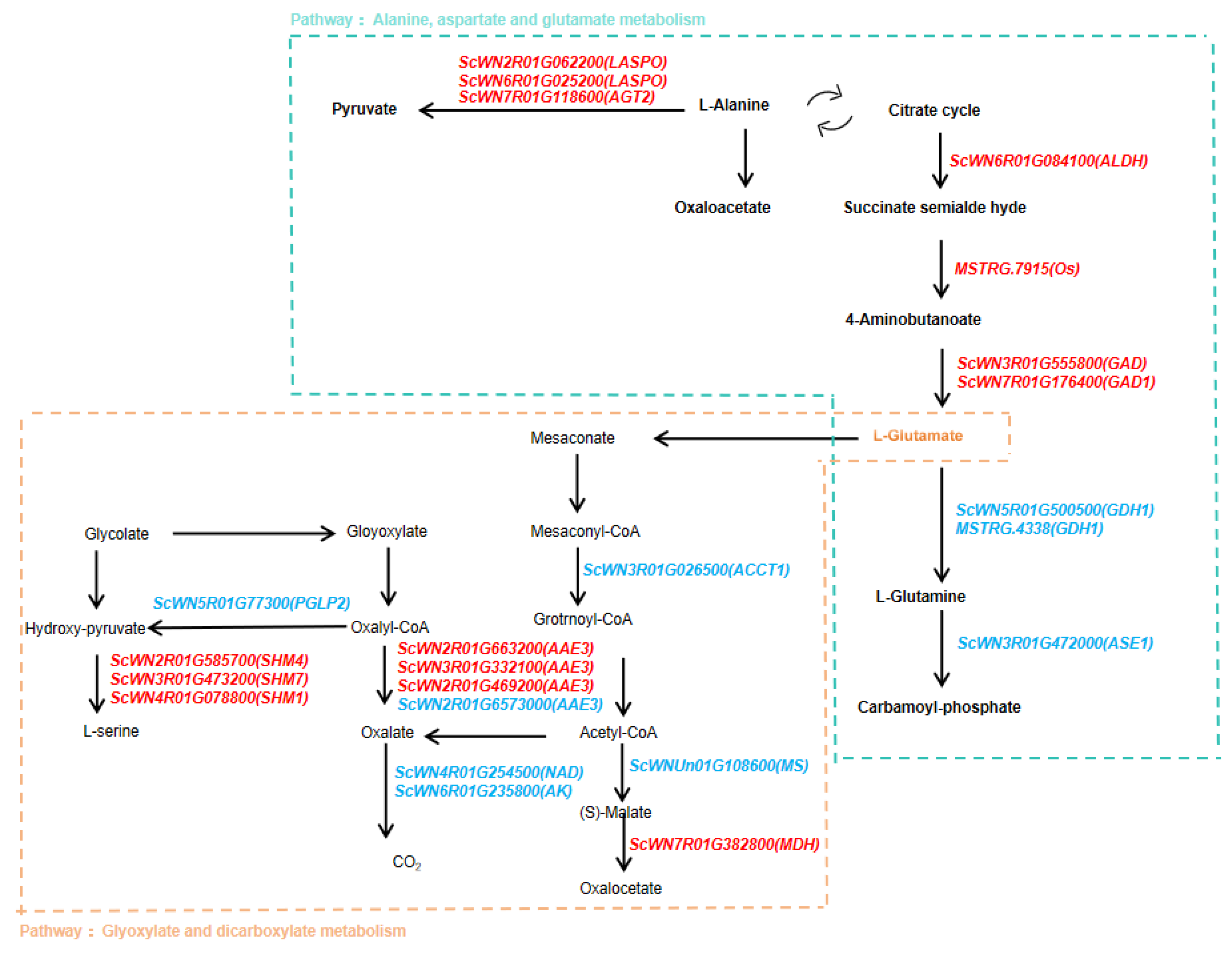
2.4. KEGG and GSEA Analysis of Differentially Expressed Genes (DEGs) in “Winter” Rye Under Cold Stress and Identification of Key Metabolic Pathways
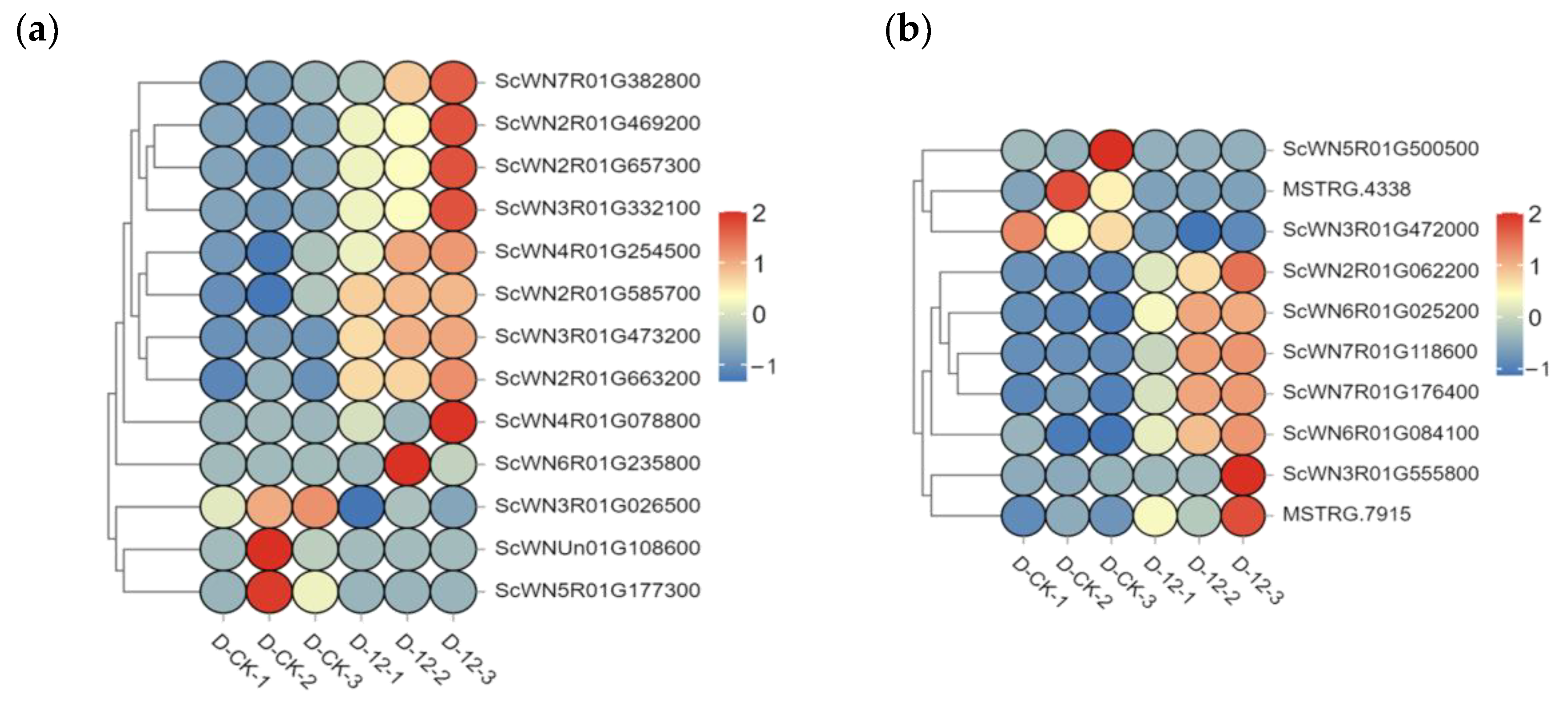
2.5. qRT-PCR Validation of Cold-Responsive Gene Expression and Expression Trends in “Winter” Rye
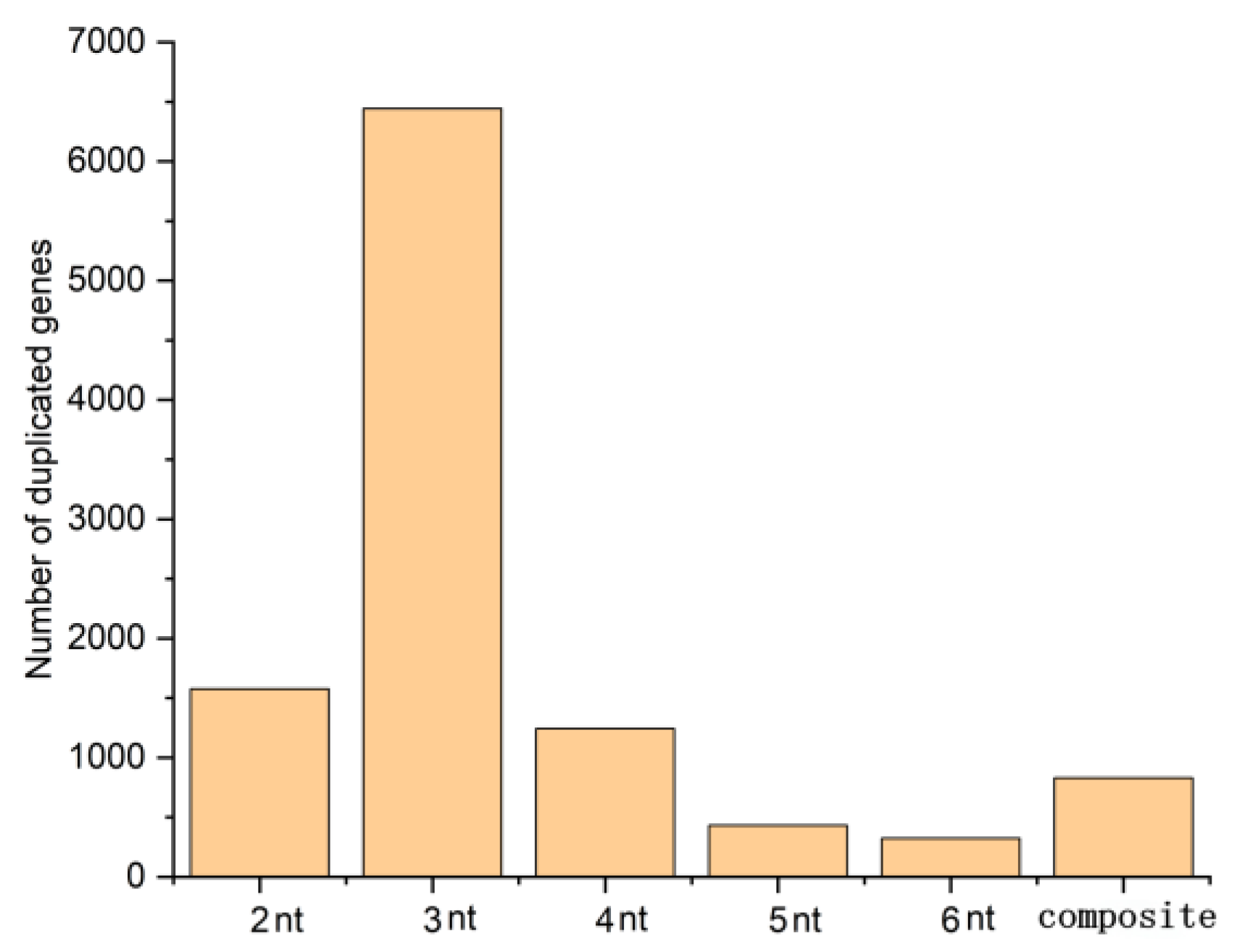
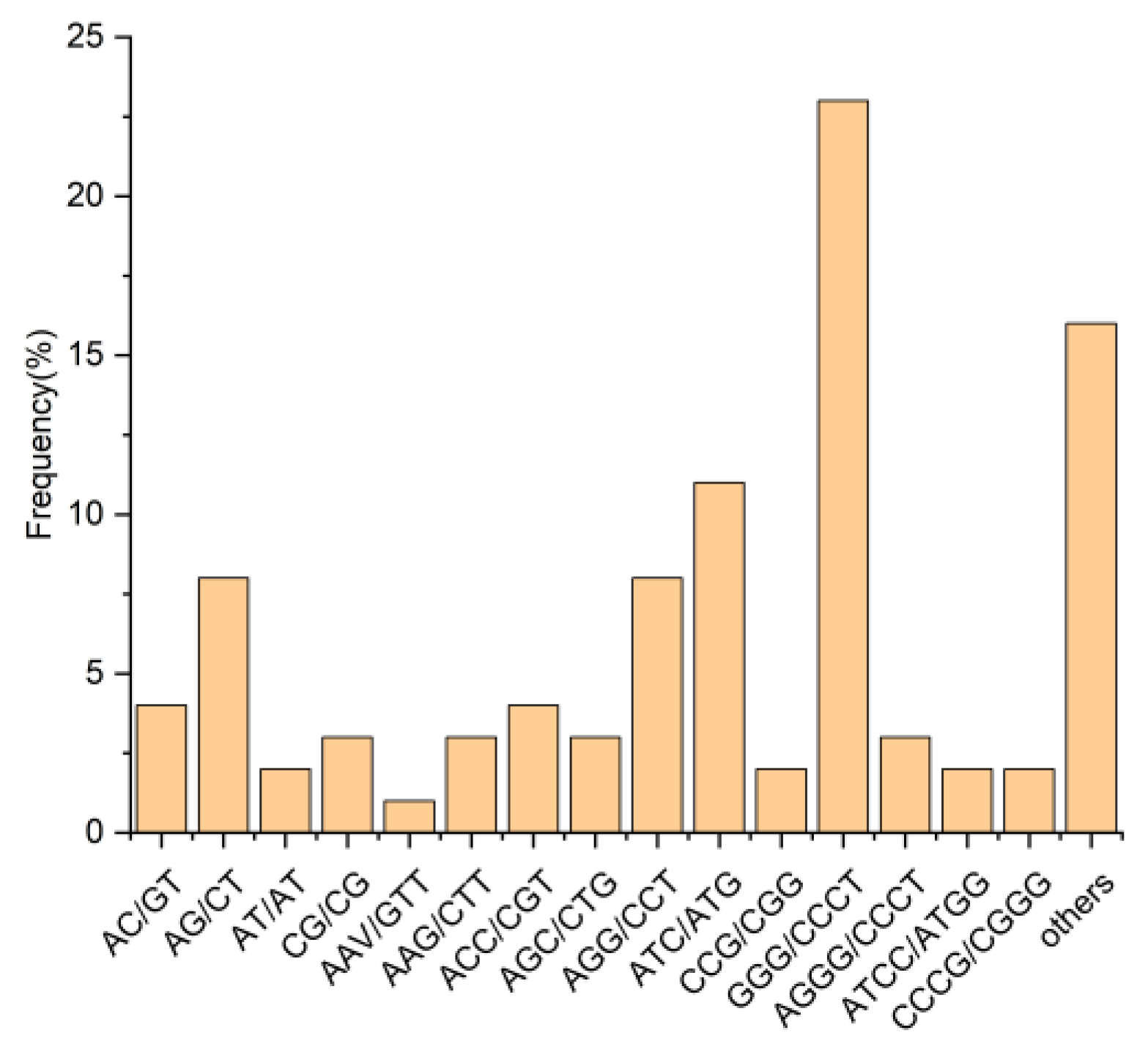
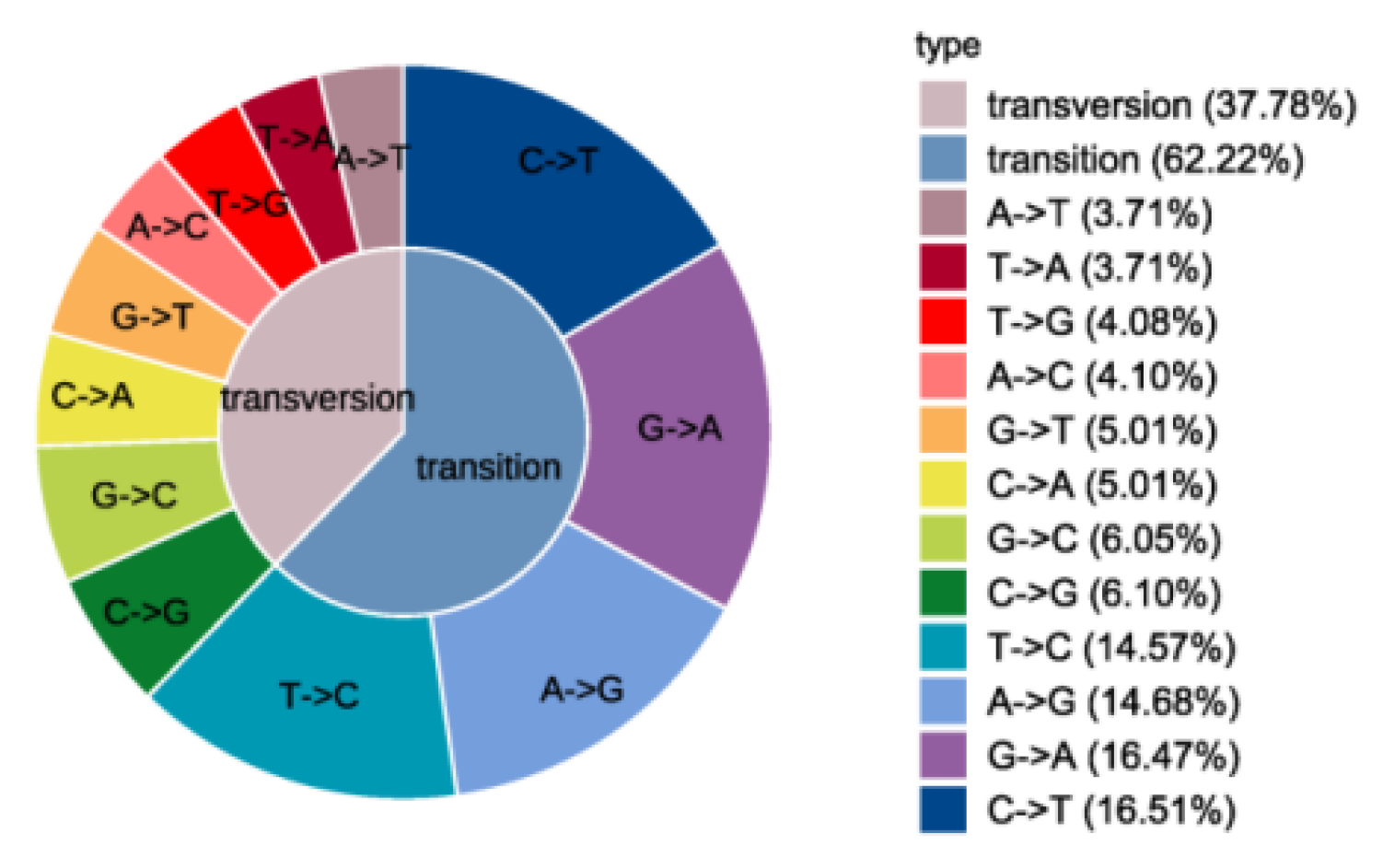
2.6. Transcriptome-Based Development and Characterization of EST-SSR and KASP Markers for Cold Tolerance in Rye
3. Materials and Methods
3.1. Plant Materials, Low-Temperature Stress Treatment, and Physiological Index Measurement
3.2. Transcriptome Sequencing
3.3. Differentially Expressed Gene (DEG) Analysis and Functional Enrichment
3.4. Gene Set Enrichment Analysis (GSEA)
3.5. qRT-PCR Validation
3.6. Development of Rye EST-SSR Molecular Markers
3.7. Development of Rye EST-SNP and KASP Molecular Markers
4. Discussion
4.1. Antioxidant Enzymes and Cold Tolerance in Plants
4.2. Role of Carbohydrate and Amino Acid Metabolism Pathways in Rye Cold Tolerance
4.3. Application of Transcriptome Sequencing and Molecular Markers in Rye Cold Tolerance Research
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | RawData | CleanData (%) | Adapter (%) | LowQuality (%) | PolyA (%) | N (%) |
|---|---|---|---|---|---|---|
| D-CK-1 | 48,707,146 | 48,582,636 (99.74%) | 13,498 (0.03%) | 111,006 (0.23%) | 0 (0.00%) | 6 (0.00%) |
| D-CK-2 | 47,871,738 | 47,739,978 (99.72%) | 14,448 (0.03%) | 117,306 (0.25%) | 0 (0.00%) | 6 (0.00%) |
| D-CK-3 | 53,268,824 | 53,127,680 (99.74%) | 15,382 (0.03%) | 125,762 (0.24%) | 0 (0.00%) | 0 (0.00%) |
| D-12-1 | 58,454,228 | 58,295,122 (99.73%) | 17,392 (0.03%) | 141,712 (0.24%) | 0 (0.00%) | 2 (0.00%) |
| D-12-2 | 62,533,210 | 62,354,096 (99.71%) | 19,844 (0.03%) | 159,258 (0.25%) | 0 (0.00%) | 12 (0.00%) |
| D-12-3 | 58,815,098 | 58,660,926 (99.74%) | 19,262 (0.03%) | 134,906 (0.23%) | 0 (0.00%) | 4 (0.00%) |
Appendix A.2
| Pathway ID | Pathway | KEGG_A Class | KEGG_B Class | Candidate Genes | Upgrade Candidate Genes | Down Candidate Genes | All Genes |
|---|---|---|---|---|---|---|---|
| ko00630 | Glyoxylate and dicarboxylate metabolism | Metabolism | Carbohydrate metabolism | 13 | 7 | 6 | 112 |
| ko00250 | Alanine, aspartate and glutamate metabolism | Metabolism | Amino acid metabolism | 10 | 4 | 6 | 79 |
Appendix A.3
| Categories | Numbers |
|---|---|
| Non-amplified band | 8 |
| Amplified band | 143 |
| Polymorphic band | 93 |
| Unitary strip | 50 |
Appendix A.4
| ID | SSR | Size | FORWARDPRIMER (5′-3′) | REVERSEPRIMER (5′-3′) | Tm (°C) | PRODUCT Size (bp) |
|---|---|---|---|---|---|---|
| Sc1R14 | (CGCCGA)4 | 24 | CCTCCTCCTCCTCCTCCTC | CCGAAGCTGTCGTAGTGGAT | 57.1 | 253 |
| Sc1R20 | (CT)7 | 14 | TCCTCCCCCTCTCTTCTCTC | ACATAACGGTCCGCAAACTC | 55.4 | 212 |
| Sc3R06 | (TTCT)6 | 24 | AGCTTCTGCTGGTCTGTGGT | AAGAATGCCAGAGCAGGCTA | 56.3 | 184 |
| Sc3R09 | (CCGC)5 | 20 | CAGCAGAGTCCAACAAACCC | AGCTGGGCGAGCTCTTGT | 56.7 | 205 |
| Sc4R02 | (CCT)5 | 15 | AACCTCGCTCCACCTCATCT | AATGGAAGGTGTCCGGGT | 56.6 | 278 |
| Sc4R08 | (TA)6 | 12 | GAGAGCCGTCACTTTTGGTC | TCACAGCAGAGATGAGGTCG | 56.3 | 165 |
| Sc4R18 | (TG)8 | 16 | AGCAAGCTGCTTCTCTGAGG | TTTGCTCACGACTTTGGTTG | 53.4 | 244 |
| Sc6R15 | (CAG)7 | 21 | ATCCAATGAATCAAGCCAGC | CATGGAGTGATGGTTAGGGG | 53 | 278 |
| Sc7R03 | (GAA)7 | 21 | GGGACTCTGACAACGACGAT | TAATGGTGGCCCAAGAAGAC | 54.8 | 148 |
| Sc1R02 | (GAT)6 | 18 | CTGGTGATACAGCCAACCCT | CGGATAGCACGGAGTACGTT | 57 | 205 |
| Sc1R03 | (TTTC)5 | 20 | AGGTTGTCATTTTCATGCCC | GAGCTGATCTTCAAGGCCAC | 53.1 | 237 |
| Sc4R04 | (ACG)9 | 27 | CTCCTCAAGTCCAGTGCTCC | TCCATCAGAGACAGCAGACG | 56.8 | 233 |
| Sc5R03 | (GC)6 | 12 | CAAATTTAGCCACCTCCCAA | CCTTTCTTTCCGCTTCCTCT | 52.6 | 247 |
Appendix A.5
| Categories | Numbers |
|---|---|
| Non-amplified band | 56 |
| Amplified band | 84 |
| Polymorphic band | 54 |
| Unitary strip | 30 |
Appendix A.6
| ID | Ref | Mut | FORWARD PRIMER1 (5′-3′) | REVERSE PRIMER1 (5′-3′) | Primer Common |
|---|---|---|---|---|---|
| KSAP-063 | C | T | GCCGCGCCGGTGA | CGCCGCGCCGGTA | AGGAGAAGGTCGGGG AGGTGAA |
| KASP-050 | C | A | CGCGGCTTGTGCAGCAGAC | CTCGCGGCTTGTGCAGCAGAA | GCAGCTTGGGGACATCGCCAAT |
| KASP-109 | G | C | CGCCGGCGTGGCG | CGCCGGCGTGGCC | GATGTCGGCCTGGATCTTGCTGTA |
| KASP-126 | T | C | CCACGCCATCGTCCTCGGT | CACGCCATCGTCCC GGC | CACTGGAGGAAGGTGCCGATGAA |
| KASP-424 | A | G | GCCCCCAGCATCTTCGCCA | CCCCCAGCATCTTG CCG | TGATGAGCCACGACCCCCTGAA |
| KASP-396 | C | T | CGGTGCACGCGCC GGC | CCGGTGCACGCGCC GGT | CCAAGCAGGCCCTTCTTCTCCTT |
| KASP-665 | G | A | GCCGCCGGTGCG GGC | GGCCGCCGGTGCG GGT | AGGAGAAGCTGCCTGGCCACAA |
Appendix A.7
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| TaActin | ATGGCTGGAGAAGGATTC | CTCCTTAATGTCACGCACG |
Appendix A.8
| Number | The Experimental Materials |
|---|---|
| 1 | “Winter” |
| 2 | HZHM3 |
| 3 | HZHM8 |
| 4 | “Victory” |
| 5 | RUS1060 HZHM132 |
| 6 | RUS1063 HZHM135 |
| 7 | RUS1087 HZHM159 |
| 8 | RUS1095 HZHM167 |
| 9 | RUS1080 HZHM158 |
| 10 | RUS110 HZHM72 |
| 11 | RUS960 HZHM32 |
| 12 | RUS1079 HZHM151 |
| 13 | RUS1081 HZHM153 |
| 14 | RUS169 |
| 15 | RUS1082 |
| 16 | RUS1064 |
| 17 | RUS1091 |
| 18 | RUS966 |
| 19 | RUS1071 |
| 20 | RUS1098 |
| 21 | RUS1075 |
| 22 | RUS1090 |
| 23 | RUS964 |
| 24 | RUS920 |
| 25 | RUS1057 |
| 26 | RUS1073 |
| 27 | RUS1070 |
| 28 | RUS1092 |
| 29 | RUS961 |
| 30 | RUS990 |
| 31 | RUS1088 |
Appendix B

References
- Rihan, H.Z.; Al-Issawi, M.; Fuller, M.P. Advances in physiological and molecular aspects of plant cold tolerance. J. Plant Interact. 2017, 12, 143–157. [Google Scholar] [CrossRef]
- Ding, Y.; Yang, S. Surviving and thriving: How plants perceive and respond to temperature stress. Dev. Cell 2022, 57, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Sun, S.; Zhao, W.; Yang, X.; Chen, Z.; Mao, H.; Sheng, L. Comprehensive physiology and proteomics analysis revealed the resistance mechanism of rice (Oryza sativa L.) to cadmium stress. Ecotoxicol. Environ. Saf. 2024, 278, 116413. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, G.; Liu, F.; Zhu, J. Responses of Antioxidant Enzymes to Chronic Free-Air Ozone Stress in Rice (Oryza sativa L.) Cultivars with Different Ozone-Sensitivities. Bull. Environ. Contam. Toxicol. 2019, 103, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Mougiou, N.; Baalbaki, B.; Doupis, G.; Kavroulakis, N.; Poulios, S.; Vlachonasios, K.E.; Koubouris, G.C. The Effect of Low Temperature on Physiological, Biochemical and Flowering Functions of Olive Tree in Relation to Genotype. Sustainability 2020, 12, 10065. [Google Scholar] [CrossRef]
- Chen, H.; Liang, X.; Yang, Z. Effects of Low-Temperature Stress on Physiological Characteristics and Microstructure of Stems and Leaves of Pinus massoniana L. Plants 2024, 13, 2229. [Google Scholar] [CrossRef]
- Wang, Y.; Xiang, C.; Li, D.; Du, M.; Mo, Y.; Jiang, Z.; Wang, J. Identification of key genes involved in the two lignifications in stored king oyster mushrooms (Pleurotus eryngii) and their expression features. Postharvest Biol. Technol. 2020, 169, 111289. [Google Scholar] [CrossRef]
- Deng, X.; Wei, Y.; Chen, Y.; Ding, P.; Xu, F.; Shao, X. Gene co-expression network analysis of the transcriptome identifies cold-resistant hub genes related to carbohydrate metabolism in peach fruit. Postharvest Biol. Technol. 2025, 220, 113300. [Google Scholar] [CrossRef]
- Erath, W.; Bauer, E.; Fowler, D.B.; Gordillo, A.; Korzun, V.; Ponomareva, M.; Schmidt, M.; Schmiedchen, B.; Wilde, P.; Schön, C.-C. Exploring new alleles for frost tolerance in winter rye. Theor. Appl. Genet. 2017, 130, 2151–2164. [Google Scholar] [CrossRef]
- Jiang, G.; Hassan, M.A.; Muhammad, N.; Arshad, M.; Chen, X.; Xu, Y.; Xu, H.; Ni, Q.; Liu, B.; Yang, W.; et al. Comparative Physiology and Transcriptome Analysis of Young Spikes in Response to Late Spring Coldness in Wheat (Triticum aestivum L.). Front. Plant Sci. 2022, 13, 811884. [Google Scholar] [CrossRef]
- Tyagi, W.; Yumnam, J.S.; Sen, D.; Rai, M. Root transcriptome reveals efficient cell signaling and energy conservation key to aluminum toxicity tolerance in acidic soil adapted rice genotype. Sci. Rep. 2020, 10, 4580. [Google Scholar] [CrossRef] [PubMed]
- Xuhui, L.; Weiwei, C.; Siqi, L.; Junteng, F.; Hang, Z.; Xiangbo, Z.; Yongwen, Q. Full-length transcriptome analysis of maize root tips reveals the molecular mechanism of cold stress during the seedling stage. BMC Plant Biol. 2022, 22, 398. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, D.; Yang, Q.; Zeng, J.; Jin, G.; Chen, Z.H.; Zhang, G.; Dai, F. Identification of Mild Freezing Shock Response Pathways in Barley Based on Transcriptome Profiling. Front. Plant Sci. 2016, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Xinquan, Y.; Peng, L.; Han, Z.; Zhongfu, N.; Sun, Q. Genetic diversity revealed by genomic-SSR and EST-SSR markers among common wheat, spelt and compactum. Prog. Nat. Sci. 2005, 15, 24–33. [Google Scholar] [CrossRef]
- Qu, K.; Wang, J.; Cheng, Y.; Bai, B.; Xia, X.; Geng, H. Identification of quantitative trait loci and candidate genes for grain superoxide dismutase activity in wheat. BMC Plant Biol. 2024, 24, 716. [Google Scholar] [CrossRef]
- Sedaghat, M.; Hazrati, S.; Omrani, M. Use of zeolite and salicylic acid as an adaptation strategy against drought in wheat plants. S. Afr. J. Bot. 2022, 146, 111–117. [Google Scholar] [CrossRef]
- Romero, I.; Teresa Sanchez-Ballesta, M.; Maldonado, R.; Isabel Escribano, M.; Merodio, C. Anthocyanin, antioxidant activity and stress-induced gene expression in high CO2-treated table grapes stored at low temperature. J. Plant Physiol. 2008, 165, 522–530. [Google Scholar] [CrossRef]
- Guy, C. Cold Acclimation And Freezing Stress Tolerance: Role Of Protein Metabolism. Annu. Rev. Plant Physiol. 1990, 41, 187–223. [Google Scholar] [CrossRef]
- Gopalakrishnan Nair, P.M.; Kang, I.-S.; Moon, B.-Y.; Lee, C.-H. Effects of low temperature stress on rice (Oryzasativa L.) plastid ω-3 desaturase gene, OsFAD8 and its functional analysis using T-DNA mutants. Plant Cell Tissue Organ Cult. (PCTOC) 2009, 98, 87–96. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, M.; Xu, K.; Li, J.; Li, S.; Zhang, S.; Yang, X. Integrated transcriptomics and metabolomics analyses provide insights into cold stress response in wheat. Crop J. 2019, 7, 857–866. [Google Scholar] [CrossRef]
- Qiao, Y.; Wu, L.; Yang, S.; Wang, Q.; Gu, H.; Wei, L.; Liu, G.; Zhou, S.; Wang, P.; Song, M. Metabolomic and transcriptomic analyses provide insights into variations in flavonoids contents between two Artemisia cultivars. BMC Plant Biol. 2023, 23, 288. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Li, C.; Zhang, Y.; Wu, Z.; Li, W.; Zhang, Z.; Ye, W.; Guo, H.; Wang, C.; Long, T.; et al. Integrated Transcriptomic and Metabolomic Analyses Reveal the Molecular and Metabolic Basis of Flavonoids in Areca catechu L. J. Agric. Food Chem. 2023, 71, 4851–4862. [Google Scholar] [CrossRef]
- Li, H.; Zhao, J.; Zhang, W.; He, T.; Meng, D.; Lu, Y.; Zhou, S.; Wang, X.; Zhao, H. Comparative Transcriptome Analysis of Two Types of Rye Under Low-Temperature Stress. Curr. Issues Mol. Biol. 2025, 47, 171. [Google Scholar] [CrossRef]
- Li, Y.; Böck, A.; Haseneyer, G.; Korzun, V.; Wilde, P.; Schön, C.-C.; Ankerst, D.P.; Bauer, E. Association analysis of frost tolerance in rye using candidate genes and phenotypic data from controlled, semi-controlled, and field phenotyping platforms. BMC Plant Biol. 2011, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Molla, K.A.; Debnath, A.B.; Ganie, S.A.; Mondal, T.K. Identification and analysis of novel salt responsive candidate gene based SSRs (cgSSRs) from rice (Oryza sativa L.). BMC Plant Biol. 2015, 15, 122. [Google Scholar] [CrossRef]
- Hwarari, D.; Guan, Y.; Ahmad, B.; Movahedi, A.; Min, T.; Hao, Z.; Lu, Y.; Chen, J.; Yang, L. ICE-CBF-COR Signaling Cascade and Its Regulation in Plants Responding to Cold Stress. Int. J. Mol. Sci. 2022, 23, 1549. [Google Scholar] [CrossRef]
- Singh, K.; Gulyás, Z.; Athmer, B.; Kovács, B.; Mednyánszky, Z.; Galiba, G.; Stein, N.; Simon-Sarkadi, L.; Kocsy, G. Comparative study of free amino acids at metabolite and gene expression levels in Triticeae during cold acclimation. J. Plant Biochem. Biotechnol. 2024, 33, 558–569. [Google Scholar] [CrossRef]
- Rahman, F.U.; Zhu, Q.; Zhang, K.; Kang, X.; Wang, X.; Chen, W.; Li, X.; Zhu, X. Transcriptome and metabolome analyses provide insights into the fruit softening disorder of papaya fruit under postharvest heat stress. Food Chem. 2024, 460, 140771. [Google Scholar] [CrossRef]
- Liu, J.; Bao, Y.; Liu, S.; Zhu, L.; Xu, X.; Jiang, G.; Zhang, Z. Physiological and transcriptomic analyses reveal mechanisms of exogenous strigolactones to regulate cold tolerance in litchi fruit. Postharvest Biol. Technol. 2024, 210, 112764. [Google Scholar] [CrossRef]
- Asakura, H.; Yamakawa, T.; Tamura, T.; Ueda, R.; Taira, S.; Saito, Y.; Abe, K.; Asakura, T. Transcriptomic and Metabolomic Analyses Provide Insights into the Upregulation of Fatty Acid and Phospholipid Metabolism in Tomato Fruit under Drought Stress. J. Agric. Food Chem. 2021, 69, 2894–2905. [Google Scholar] [CrossRef]
- Song, Z.; Yan, A.; Li, Z.; Shang, Y.; Chen, R.; Yang, Z.; Guo, Z.; Zhang, Y.; Wen, T.; Ogaji, O.D.; et al. Integrated metabolomic and transcriptomic analysis reveals the effects and mechanisms of Jinqi Jiangtang tablets on type 2 diabetes. Phytomedicine 2024, 134, 155957. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Muthusamy, S.K.; Singh, G.P.; Sharma, P. Identification and development of novel salt-responsive candidate gene based SSRs (cg-SSRs) and MIR gene based SSRs (mir-SSRs) in bread wheat (Triticum aestivum). Sci. Rep. 2021, 11, 2210. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Jia, J.; Kong, X. A SNP-Based Molecular Barcode for Characterization of Common Wheat. PLoS ONE 2016, 11, e0150947. [Google Scholar] [CrossRef]
- Yang, F.; Lang, T.; Wu, J.; Zhang, C.; Qu, H.; Pu, Z.; Yang, F.; Yu, M.; Feng, J. SNP loci identification and KASP marker development system for genetic diversity, population structure, and fingerprinting in sweetpotato (Ipomoea batatas L.). BMC Genom. 2024, 25, 1245. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Lin, J.; Wang, Y.; An, H.; Chen, H.; Pan, G.; Zhang, S.; Guo, B.; Yu, K.; Li, H.; et al. Selection and Validation of 48 KASP Markers for Variety Identification and Breeding Guidance in Conventional and Hybrid Rice (Oryza sativa L.). Rice 2022, 15, 48. [Google Scholar] [CrossRef]
- Fang, T.; Lei, L.; Li, G.; Powers, C.; Hunger, R.M.; Carver, B.F.; Yan, L. Development and deployment of KASP markers for multiple alleles of Lr34 in wheat. Theor. Appl. Genet. 2020, 133, 2183–2195. [Google Scholar] [CrossRef]
- Liu, G.; Mullan, D.; Zhang, A.; Liu, H.; Liu, D.; Yan, G. Identification of KASP markers and putative genes for pre-harvest sprouting resistance in common wheat (Triticum aestivum L.). Crop J. 2023, 11, 549–557. [Google Scholar] [CrossRef]
- Peng, X.-A.; Lu, M.-A.; Zhang, L.; Liu, T.; Cao, L.; Song, Y.-H.; Zheng, W.-Y.; He, X.-F.; Zhu, Y.-L. Genome-wide association study of major grain quality traits in wheat based on 55K SNP arrays. Acta Agron. Sin. 2024, 50, 1948–1960. [Google Scholar]
- Hasanuzzaman, M.; Saha, N.R.; Farabi, S.; Tahjib-Ul-Arif, M.; Yasmin, S.; Haque, M.S. Screening of salt-tolerant wheat (Triticum aestivum L.) through morphological and molecular markers. Cereal Res. Commun. 2023, 51, 87–100. [Google Scholar] [CrossRef]


| Chromosome Location | Number |
|---|---|
| Chromosome1R | 1491 |
| Chromosome2R | 1628 |
| Chromosome3R | 1398 |
| Chromosome4R | 1448 |
| Chromosome5R | 1757 |
| Chromosome6R | 1374 |
| Chromosome7R | 1404 |
| Unknown Chromosome | 346 |
| Total | 10,846 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Zhao, J.; He, C.; Guan, Y.; Guan, H.; He, T.; Meng, D.; Wang, X.; Tang, Y. Integrative Physiological and Transcriptomic Analysis Reveals Metabolic Adaptation and Cold-Tolerance Marker Development in Winter Rye Under Low-Temperature Stress. Plants 2025, 14, 1588. https://doi.org/10.3390/plants14111588
Li H, Zhao J, He C, Guan Y, Guan H, He T, Meng D, Wang X, Tang Y. Integrative Physiological and Transcriptomic Analysis Reveals Metabolic Adaptation and Cold-Tolerance Marker Development in Winter Rye Under Low-Temperature Stress. Plants. 2025; 14(11):1588. https://doi.org/10.3390/plants14111588
Chicago/Turabian StyleLi, Haonan, Jiahuan Zhao, Chenguang He, Yang Guan, Huimin Guan, Ting He, Dexu Meng, Xiaoping Wang, and Yimiao Tang. 2025. "Integrative Physiological and Transcriptomic Analysis Reveals Metabolic Adaptation and Cold-Tolerance Marker Development in Winter Rye Under Low-Temperature Stress" Plants 14, no. 11: 1588. https://doi.org/10.3390/plants14111588
APA StyleLi, H., Zhao, J., He, C., Guan, Y., Guan, H., He, T., Meng, D., Wang, X., & Tang, Y. (2025). Integrative Physiological and Transcriptomic Analysis Reveals Metabolic Adaptation and Cold-Tolerance Marker Development in Winter Rye Under Low-Temperature Stress. Plants, 14(11), 1588. https://doi.org/10.3390/plants14111588