

Characterization, Expression Profile Analysis, and Functional Prediction of UGP Gene Family in Dendrocalamus brandisii

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Identification of UGP Gene Families in D. brandisii
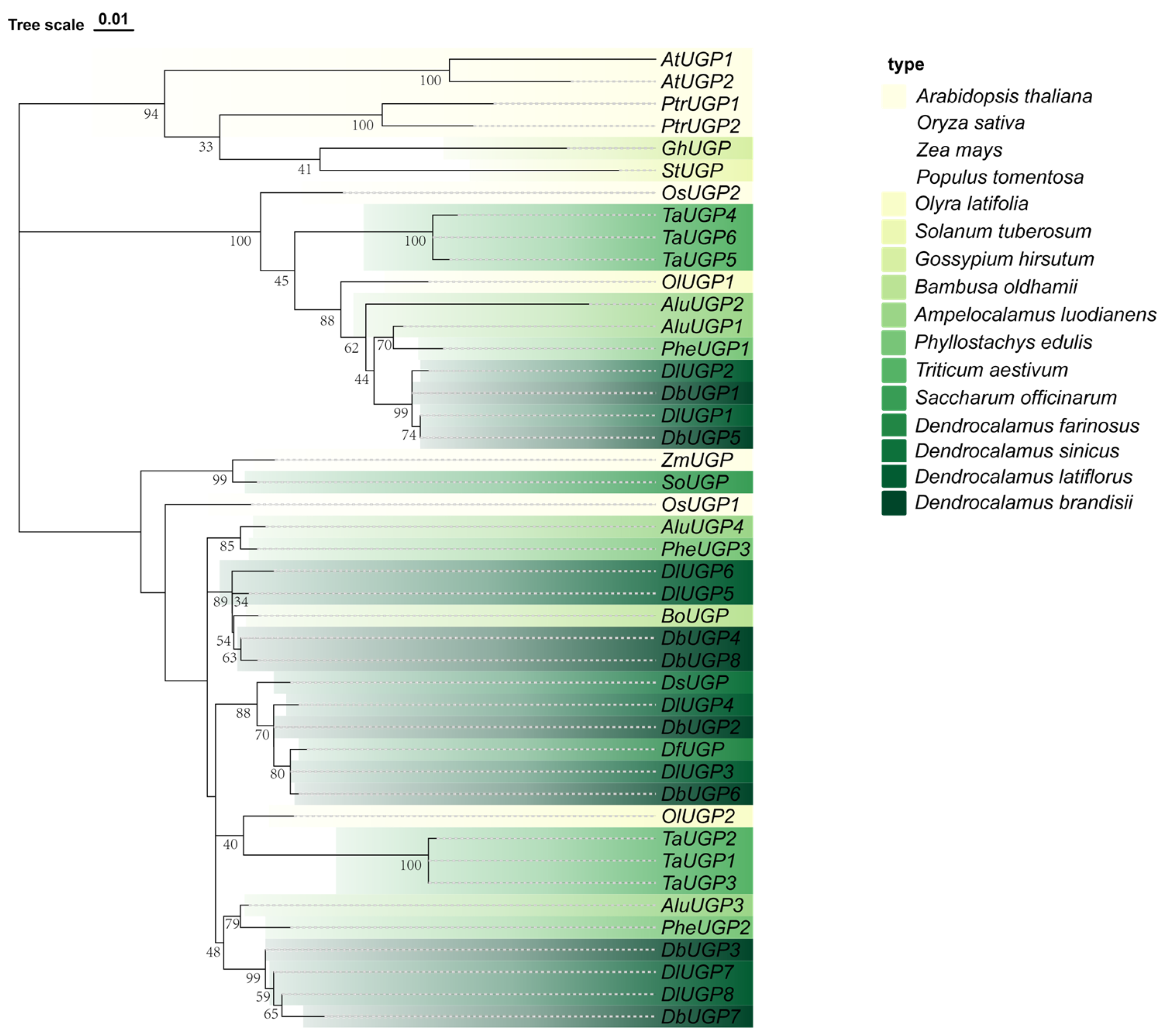
2.2. DbUGPs Were Evolutionarily Conserved and Distributed Across Two Clades with OsUGPs
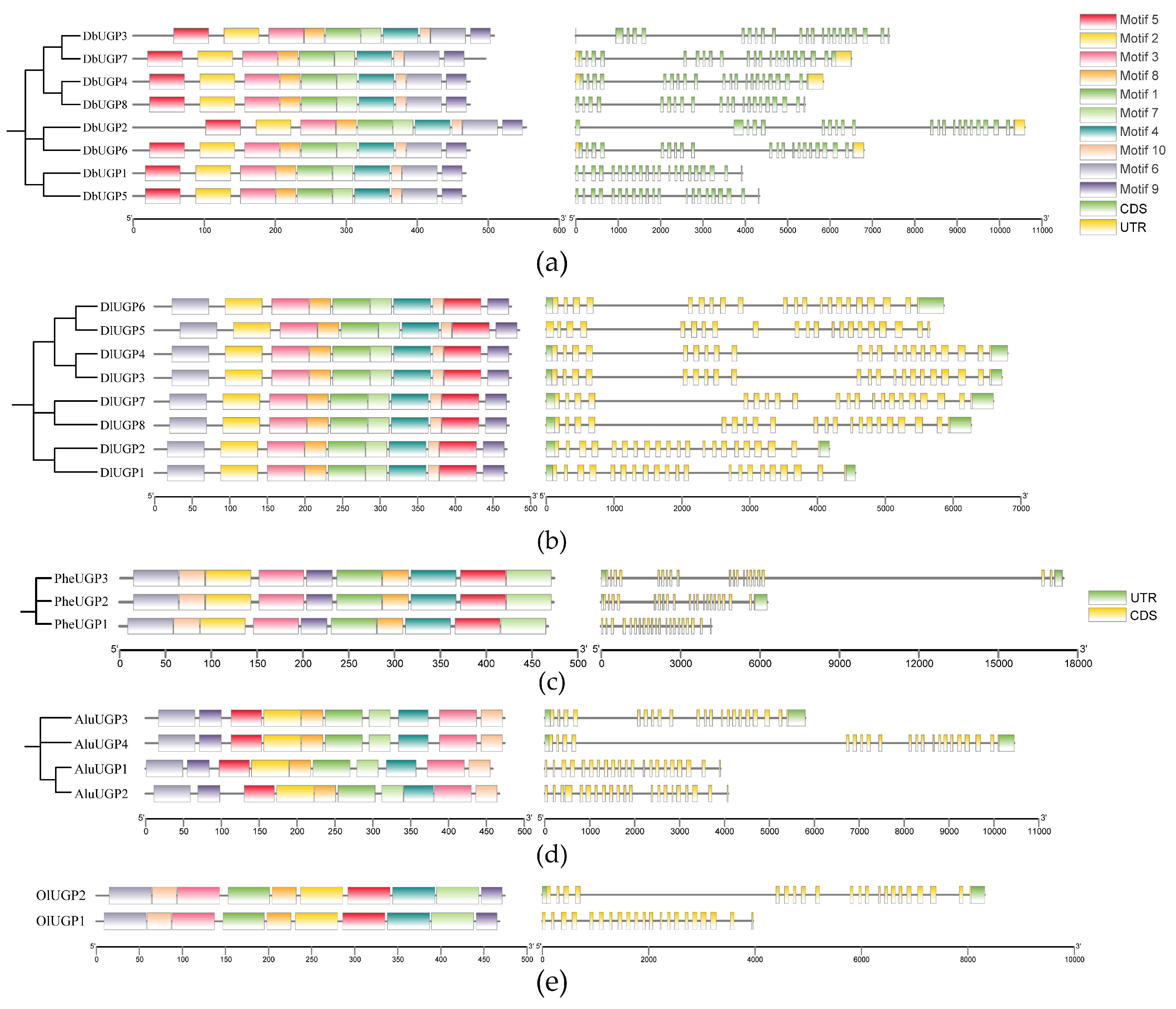
2.3. Gene Conserved Motif, Structure Analysis, and Protein Functional Domain Prediction of DbUGPs
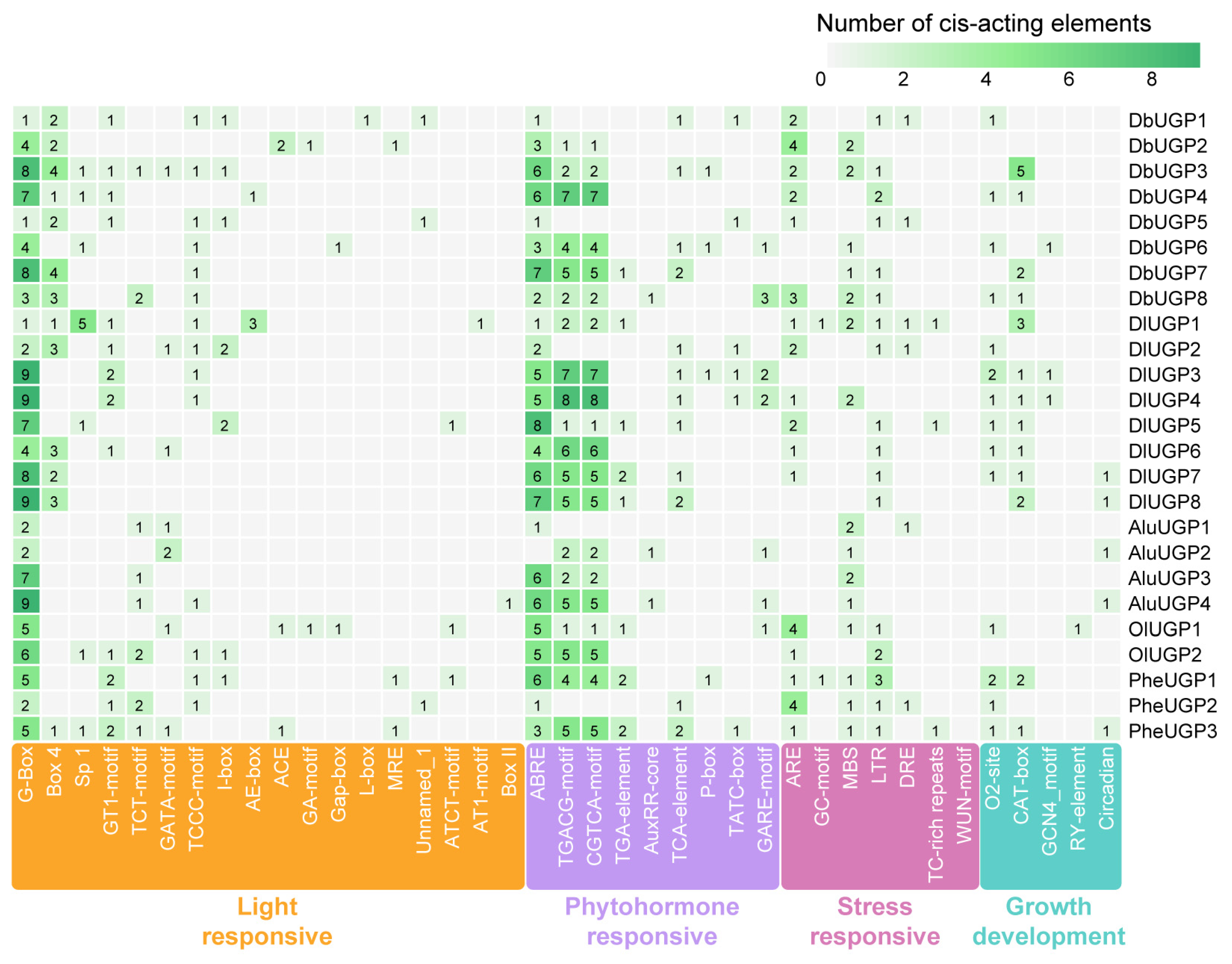
2.4. DbUGPs Promoters Mainly Responded to Light, MeJA, and ABA
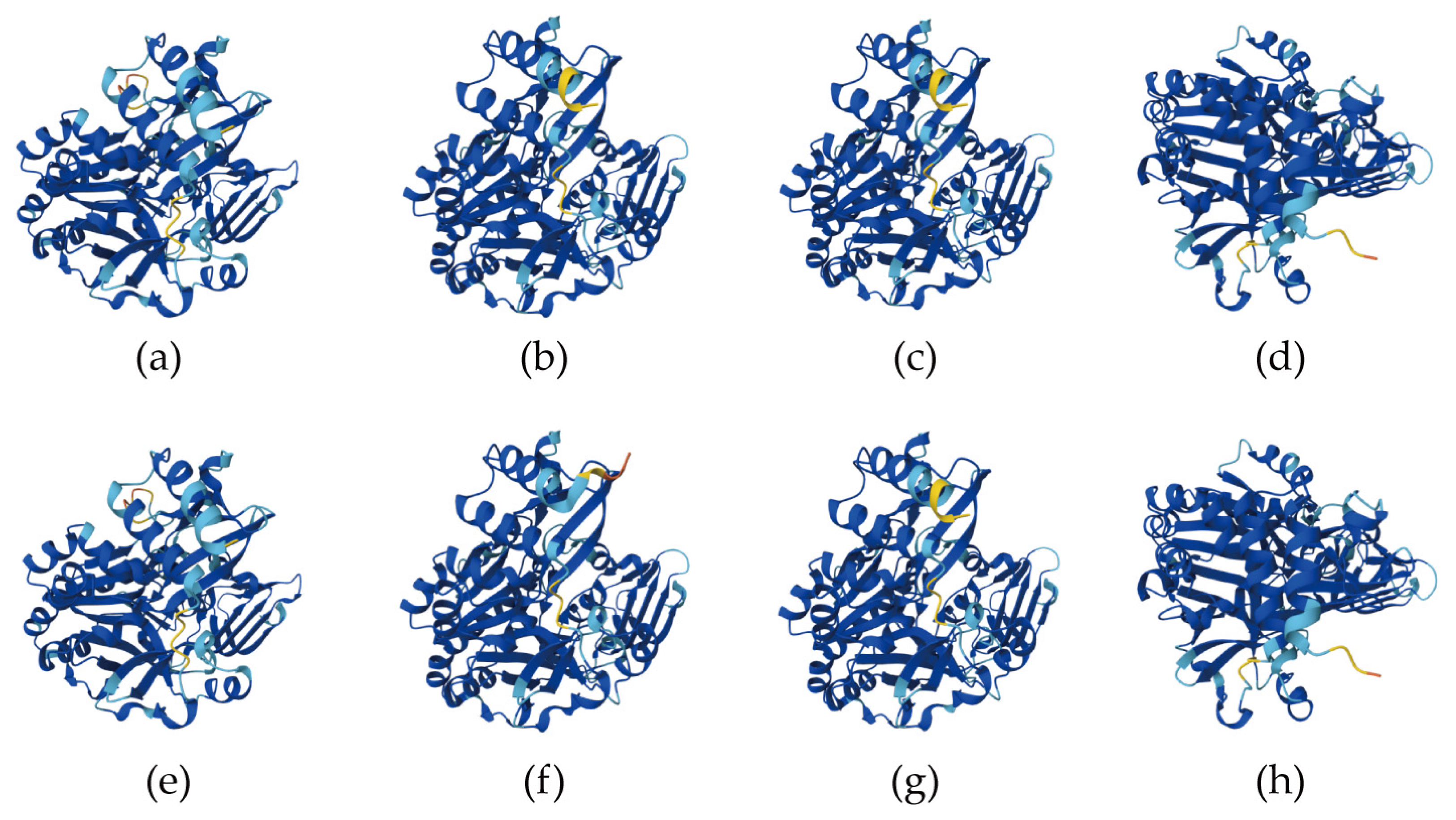
2.5. Prediction of Tertiary Structure and Secondary Structure Content in DbUGP Proteins
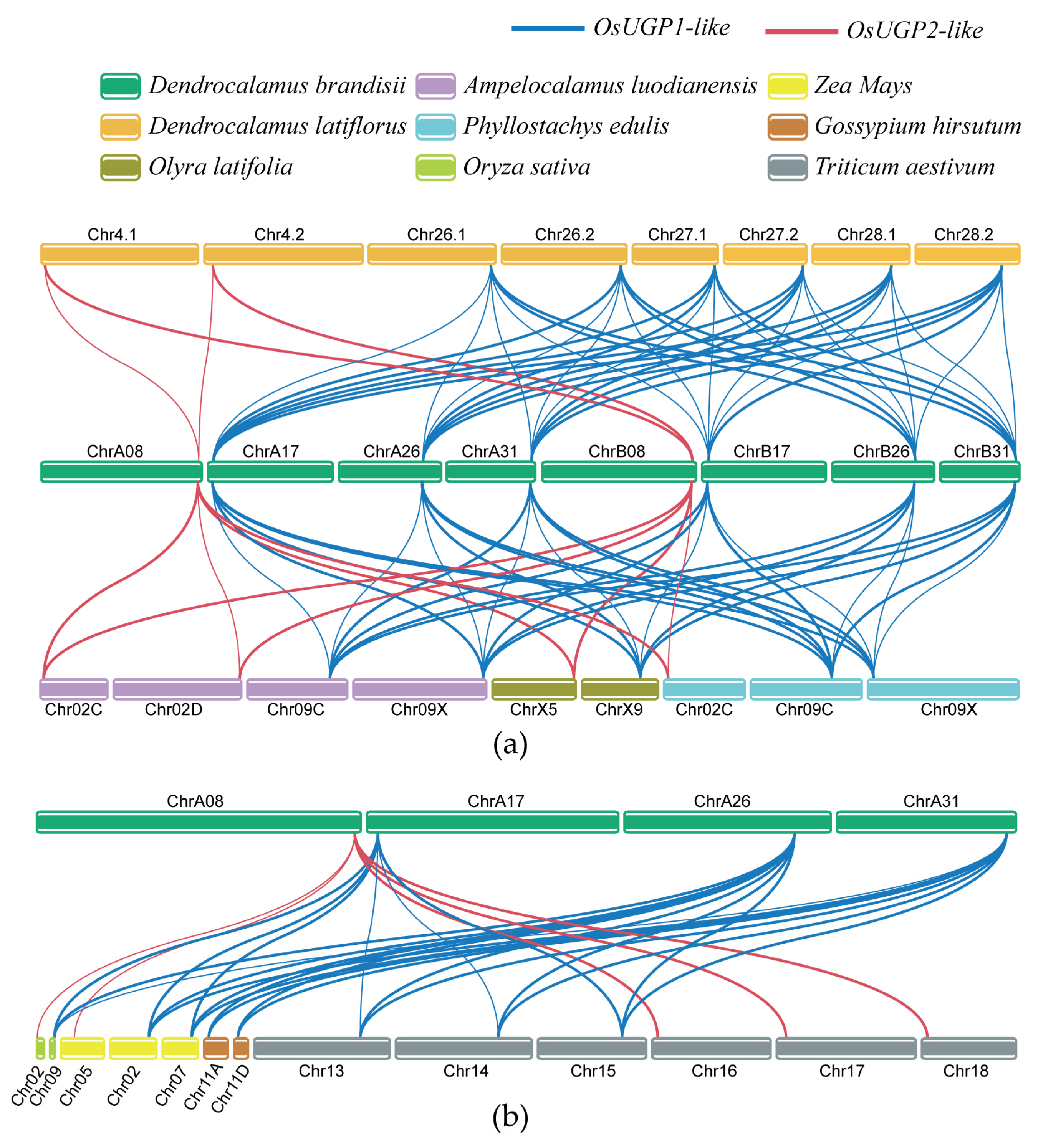
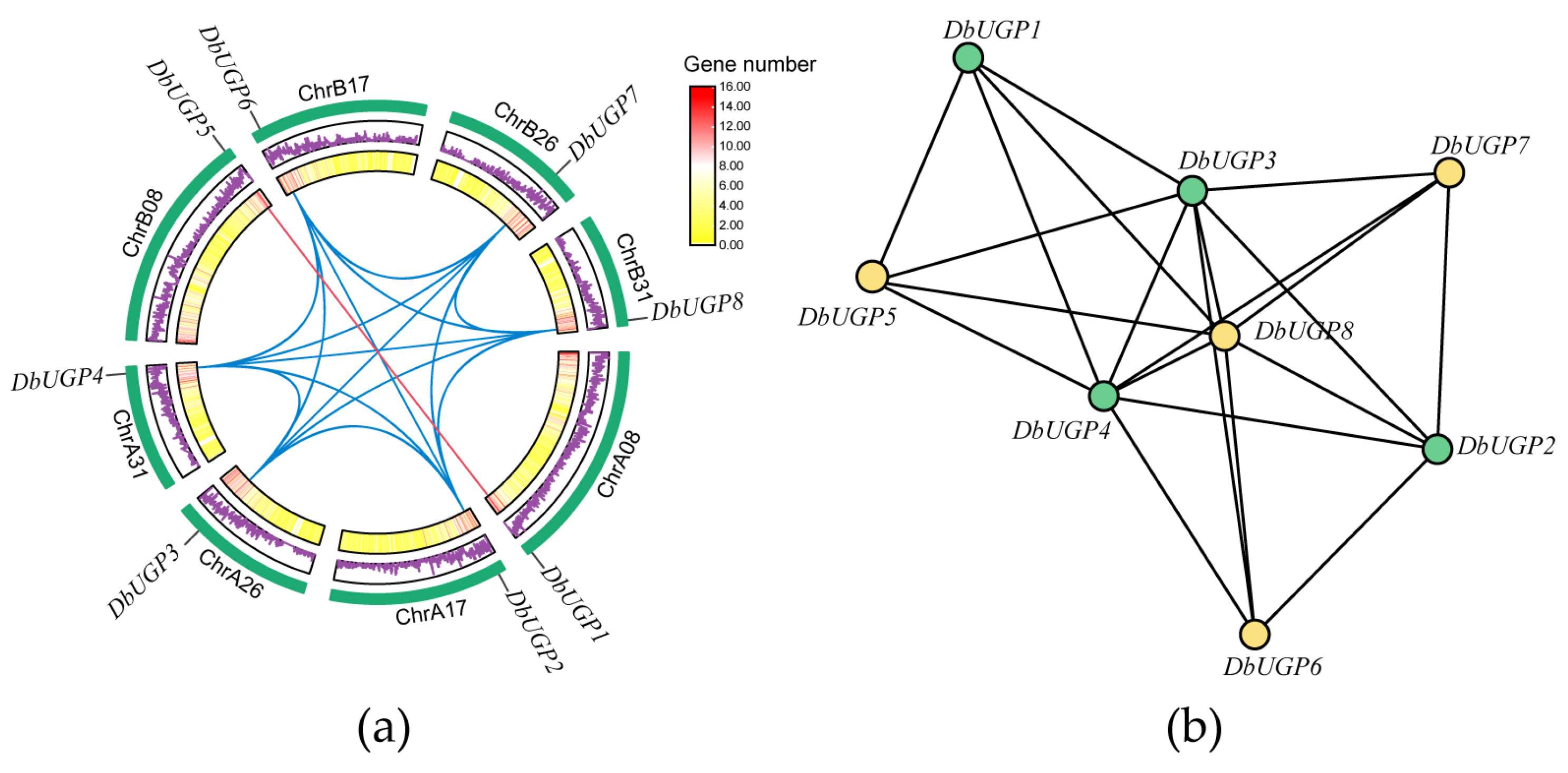
2.6. Collinearity, Evolutionary Selection Pressure, and Replication Type of DbUGPs
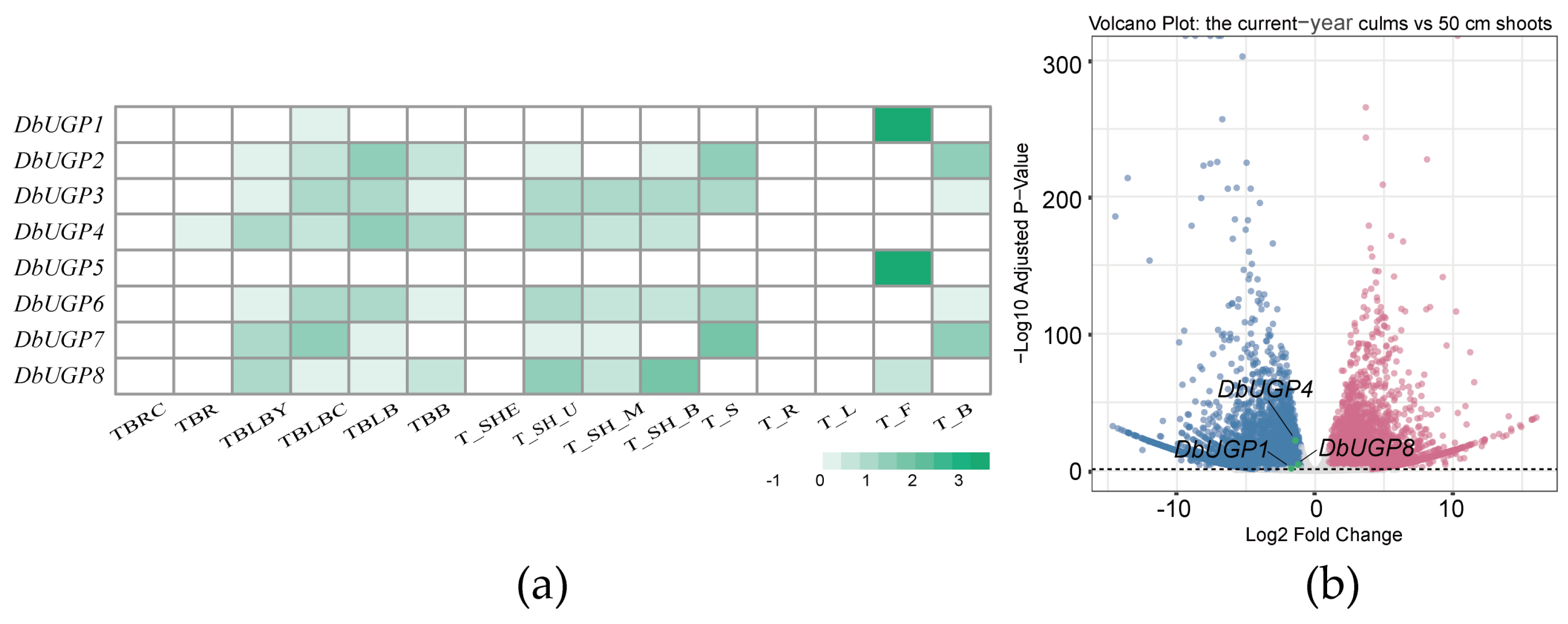
2.7. Transcriptome Analysis of DbUGPs in Different Organs and Culms at Different Developmental Stages
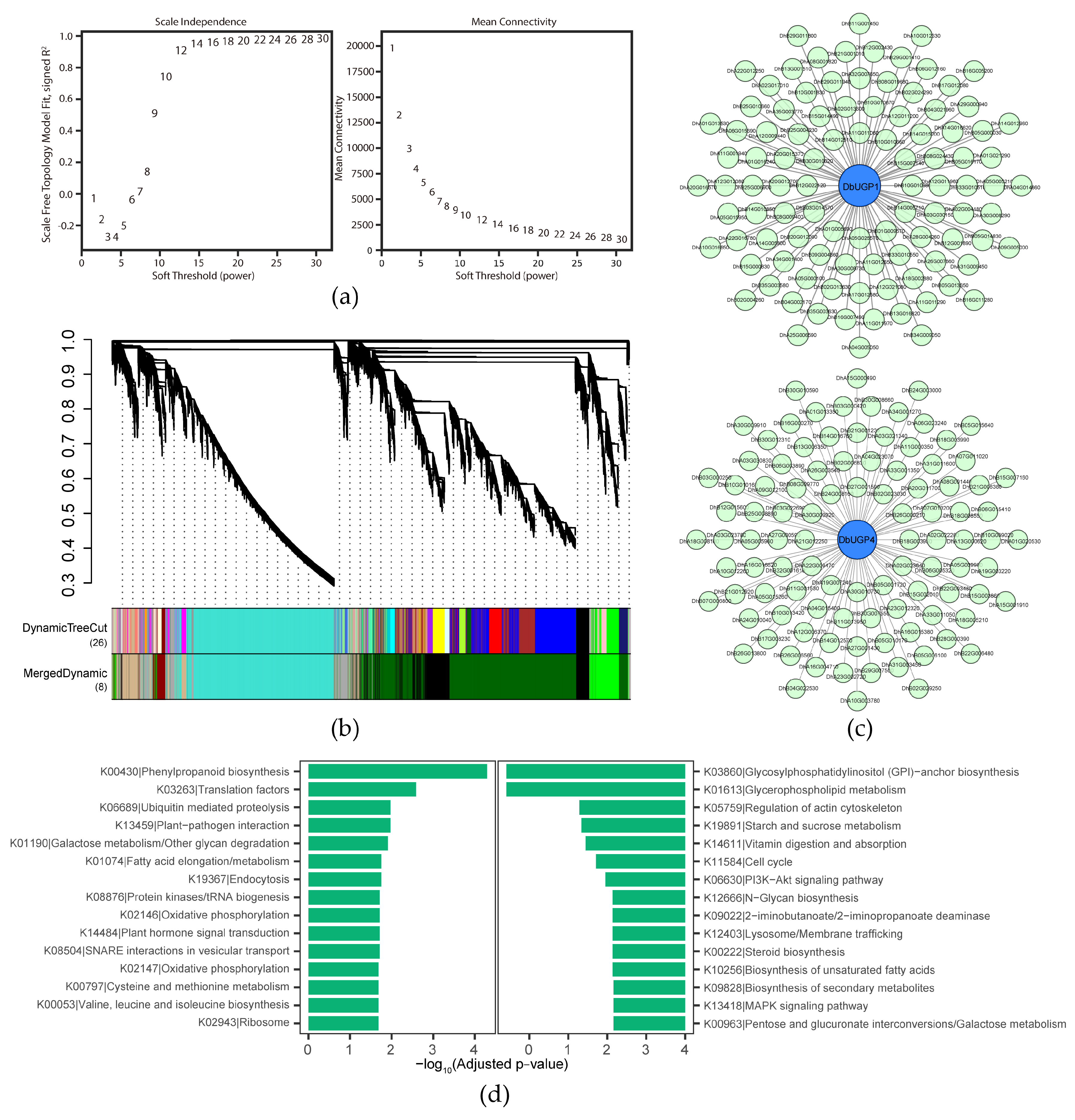
2.8. WGCNA, PPI, and KEGG Analyses Suggested DbUGP1 and DbUGP4 Involvement in Growth, Metabolism, and Stress Adaptation
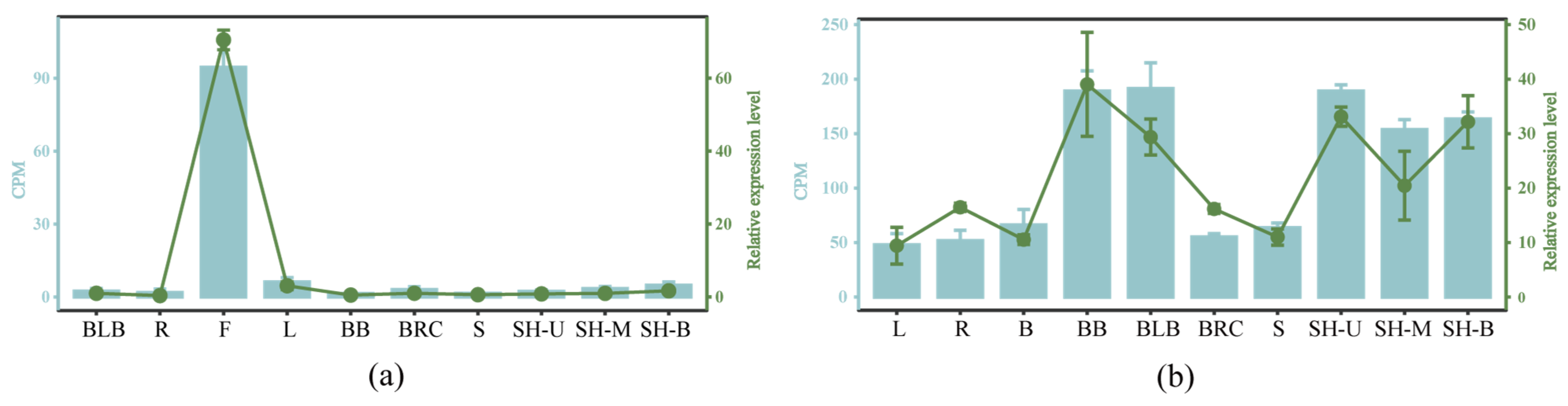
2.9. qRT-PCR Validation of Transcriptome Data from Various Tissues and Organs of D. brandisii
3. Discussion
4. Materials and Methods
4.1. Identification and Analysis of UGP Gene Families in D. brandisii
4.2. Physical and Chemical Properties of DbUGPs
4.3. Phylogenetic, Collinearity, Evolutionary Selection Pressure, and Duplication Analysis of DbUGPs
4.4. Analysis of Gene Expression Patterns
4.5. qRT-PCR Validation of the Accuracy of Transcriptome Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kleczkowski, L.A.; Decker, D. Sugar activation for production of nucleotide sugars as substrates for glycosyltransferases in plants. J. Appl. Glycosci. 2015, 62, 25–36. [Google Scholar] [CrossRef]
- Kleczkowski, L.A.; Geisler, M.; Ciereszko, I.; Johansson, H. UDP-glucose pyrophosphorylase. An old protein with new tricks. Plant Physiol. 2004, 134, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Geisler, M.; Johansson, H.; Mellerowicz, E.J.; Karpinski, S.; Kleczkowski, L.A. Differential tissue/organ-dependent expression of two sucrose-and cold-responsive genes for UDP-glucose pyrophosphorylase in Populus. Gene 2007, 389, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.H.; Zhang, X.; Li, F.G.; Hou, Y.X.; Liu, X.L.; Zhang, X.Y. Identification of a UDP-glucose pyrophosphorylase from cotton (Gossypium hirsutum L.) involved in cellulose biosynthesis in Arabidopsis thaliana. Plant Cell Rep. 2011, 30, 1303–1312. [Google Scholar] [CrossRef]
- Chen, R.Z.; Zhao, X.; Shao, Z.; Zhu, L.L.; He, G.C. Multiple isoforms of UDP-glucose pyrophosphorylase in rice. Physiol. Plant 2007, 129, 725–736. [Google Scholar] [CrossRef]
- Park, J.I.; Ishimizu, T.; Suwabe, K.; Sudo, K.; Masuko, H.; Hakozaki, H.; Nou, I.S.; Suzuki, G.; Watanabe, M. UDP-glucose pyrophosphorylase is rate limiting in vegetative and reproductive phases in Arabidopsis thaliana. Plant Cell Physiol. 2010, 51, 981–996. [Google Scholar] [CrossRef]
- Peng, Z.H.; Lu, Y.; Li, L.B.; Zhao, Q.; Feng, Q.; Gao, Z.M.; Lu, H.Y.; Hu, T.; Yao, N.; Liu, K.Y. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat. Genet. 2013, 45, 456–461. [Google Scholar] [CrossRef]
- Song, X.Z.; Peng, C.H.; Zhou, G.M.; Gu, H.H.; Li, Q.; Zhang, C. Dynamic allocation and transfer of non-structural carbohydrates, a possible mechanism for the explosive growth of Moso bamboo (Phyllostachys heterocycla). Sci. Rep. 2016, 6, 25908. [Google Scholar] [CrossRef]
- Zheng, H.F.; Bai, Y.C.; Li, X.Y.; Song, H.J.; Cai, M.M.; Cheng, Z.C.; Mu, S.H.; Li, J.; Gao, J. Photosynthesis, phytohormone signaling and sugar catabolism in the culm sheaths of Phyllostachys edulis. Plants 2022, 11, 2866. [Google Scholar] [CrossRef]
- Zhang, H.; Ying, Y.Q.; Wang, J.; Zhao, X.H.; Zeng, W.; Beahan, C.; He, J.B.; Chen, X.Y.; Bacic, A.; Song, L.L. Transcriptome analysis provides insights into xylogenesis formation in Moso bamboo (Phyllostachys edulis) shoot. Sci. Rep. 2018, 8, 3951. [Google Scholar] [CrossRef]
- Gan, X.H.; Ding, Y.L. A special long-lived cell: The culm fiber of Phyllostachys edulis (Gramineae). Plant Divers. 2008, 30, 151–158. [Google Scholar]
- Zheng, X.; Lin, S.Y.; Fu, H.J.; Wan, Y.W.; Ding, Y.L. The bamboo flowering cycle sheds light on flowering diversity. Front. Plant Sci. 2020, 11, 381. [Google Scholar] [CrossRef]
- Seethalakshmi, K.; Kumar, M.M.; Pillai, K.S.; Sarojam, N. Bamboos of India: A Compendium; Kerala Forest Research Institute & International Network for Bamboo and Rattan: Kerala, India; Beijing, China, 1998. [Google Scholar]
- Zhan, H.; Zhang, L.Y.; Deng, L.; Niu, Z.H.; Li, M.B.; Wang, C.M.; Wang, S.G. Physiological and anatomical response of foliar silicon application to Dendrocalamus brandisii plantlet leaves under chilling. Acta Physiol. Plant 2018, 40, 208. [Google Scholar] [CrossRef]
- Li, D.Z.; Xue, J.R. The Bamboos; Academic Press for the Linnean Society of London: San Diego, CA, USA, 1996; pp. 24–29. [Google Scholar]
- He, T.Y.; Zheng, J.M.; Chen, L.Y.; Rong, J.D.; Zheng, Y.S. Complete plastid genome of Dendrocalamus brandisii (Poaceae, Bambusoideae). Mitochondrial DNA Part B 2020, 5, 1286–1287. [Google Scholar] [CrossRef]
- Li, N.N.; Wang, L.; Zhang, W.B.; Takechi, K.; Takano, H.; Lin, X.F. Overexpression of UDP-glucose pyrophosphorylase from Larix gmelinii enhances vegetative growth in transgenic Arabidopsis thaliana. Plant Cell Rep. 2014, 33, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Coleman, H.D.; Ellis, D.D.; Gilbert, M.; Mansfield, S.D. Up-regulation of sucrose synthase and UDP-glucose pyrophosphorylase impacts plant growth and metabolism. Plant Biotechnol. J. 2006, 4, 87–101. [Google Scholar] [CrossRef]
- Jiang, J.T.; Zhang, Z.Y.; Bai, Y.C.; Wang, X.J.; Dou, Y.P.; Geng, R.M.; Wu, C.Y.; Zhang, H.X.; Lu, C.F.; Gu, L.F. Chromosomal-level genome and metabolome analyses of highly heterozygous allohexaploid Dendrocalamus brandisii elucidate shoot quality and developmental characteristics. J. Integr. Plant Biol. 2024, 66, 1087–1105. [Google Scholar] [CrossRef]
- Xie, J.M.; Chen, Y.R.; Cai, G.J.; Cai, R.L.; Hu, Z.; Wang, H. Tree Visualization By One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar] [CrossRef]
- Borovkov, A.Y.; McClean, P.E.; Secor, G.A. Organization and transcription of the gene encoding potato UDP-glucose pyrophosphorylase. Gene 1997, 186, 293–297. [Google Scholar] [CrossRef]
- Katsube, T.; Kazuta, Y.; Tanizawa, K.; Fukui, T. Expression in Escherichia coli of UDP-glucose pyrophosphorylase cDNA from potato tuber and functional assessment of the five lysyl residues located at the substrate-binding site. Biochemistry 1991, 30, 8546–8551. [Google Scholar] [CrossRef]
- Abe, T.; Niiyama, H.; Sasahara, T. Cloning of cDNA for UDP-glucose pyrophosphorylase and the expression of mRNA in rice endosperm. Theor. Appl. Genet. 2002, 105, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Cartharius, K.; Frech, K.; Grote, K.; Klocke, B.; Haltmeier, M.; Klingenhoff, A.; Frisch, M.; Bayerlein, M.; Werner, T. MatInspector and beyond: Promoter analysis based on transcription factor binding sites. Bioinformatics 2005, 21, 2933–2942. [Google Scholar] [CrossRef]
- Jiang, J.T.; Mu, C.H.; Bai, Y.C.; Cheng, W.L.; Geng, R.M.; Xu, J.L.; Dou, Y.P.; Cheng, Z.C.; Gao, J. Selection and validation of reference genes in Dendrocalamus brandisii for Quantitative Real-Time PCR. Plants 2024, 13, 2363. [Google Scholar] [CrossRef]
- Christopher, H.; van Rensburg, J.; Van den Ende, W. UDP-glucose: A potential signaling molecule in plants? Front. Plant Sci. 2018, 8, 2230. [Google Scholar]
- Chen, R.Z.; Zhao, X.; Shao, Z.; Wei, Z.; Wang, Y.Y.; Zhu, L.L.; Zhao, J.; Sun, M.X.; He, R.F.; He, G.C. Rice UDP-glucose pyrophosphorylase1 is essential for pollen callose deposition and its cosuppression results in a new type of thermosensitive genic male sterility. Plant Cell 2007, 19, 847–861. [Google Scholar] [CrossRef] [PubMed]
- Katsube, T.; Kazuta, Y.; Mori, H.; Nakano, K.; Tanizawa, K.; Fukui, T. UDP-glucose pyrophosphorylase from potato tuber: cDNA cloning and sequencing. J. Biochem. 1990, 108, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Pua, E.C.; Lim, S.S.W.; Liu, P.; Liu, J.Z. Expression of a UDPglucose pyrophosphorylase cDNA during fruit ripening of banana (Musa acuminata). Funct. Plant Biol. 2000, 27, 1151–1159. [Google Scholar] [CrossRef]
- Wu, L.M.; Zhang, M.L.; Zhang, R.; Yu, H.Z.; Wang, H.L.; Li, J.Y.; Wang, Y.M.; Hu, Z.; Wang, Y.T.; Luo, Z. Down-regulation of OsMYB103L distinctively alters beta-1, 4-glucan polymerization and cellulose microfibers assembly for enhanced biomass enzymatic saccharification in rice. Biotechnol. Biofuels 2021, 14, 245. [Google Scholar] [CrossRef]
- Dai, N.; Petreikov, M.; Portnoy, V.; Katzir, N.; Pharr, D.M.; Schaffer, A.A. Cloning and expression analysis of a UDP-galactose/glucose pyrophosphorylase from melon fruit provides evidence for the major metabolic pathway of galactose metabolism in raffinose oligosaccharide metabolizing plants. Plant Physiol. 2006, 142, 294–304. [Google Scholar] [CrossRef]
- Eimert, K.; Villand, P.; Kilian, A.; Kleczkowski, L.A. Cloning and characterization of several cDNAs for UDP-glucose pyrophosphorylase from barley (Hordeum vulgare) tissues. Gene 1996, 170, 227–232. [Google Scholar] [CrossRef]
- Geisler, M.; Wilczynska, M.; Karpinski, S.; Kleczkowski, L.A. Toward a blueprint for UDP-glucose pyrophosphorylase structure/function properties: Homology-modeling analyses. Plant Mol. Biol. 2004, 56, 783–794. [Google Scholar] [CrossRef] [PubMed]
- McCoy, J.G.; Bitto, E.; Bingman, C.A.; Wesenberg, G.E.; Bannen, R.M.; Kondrashov, D.A.; Phillips Jr, G.N. Structure and Dynamics of UDP–Glucose Pyrophosphorylase from Arabidopsis thaliana with Bound UDP–Glucose and UTP. J. Mol. Biol. 2007, 366, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Kleczkowski, L.A.; Kunz, S.; Wilczynska, M. Mechanisms of UDP-glucose synthesis in plants. Crit. Rev. Plant Sci. 2010, 29, 191–203. [Google Scholar] [CrossRef]
- McFarlane, H.E.; Döring, A.; Persson, S. The cell biology of cellulose synthesis. Annu. Rev. Plant Biol. 2014, 65, 69–94. [Google Scholar] [CrossRef]
- Long, W.H.; Dong, B.N.; Wang, Y.H.; Pan, P.Y.; Wang, Y.L.; Liu, L.L.; Chen, X.L.; Liu, X.; Liu, S.J.; Tian, Y.L. FLOURY ENDOSPERM8, encoding the UDP-glucose pyrophosphorylase 1, affects the synthesis and structure of starch in rice endosperm. J. Plant Biol. 2017, 60, 513–522. [Google Scholar] [CrossRef]
- Okazaki, Y.; Shimojima, M.; Sawada, Y.; Toyooka, K.; Narisawa, T.; Mochida, K.; Tanaka, H.; Matsuda, F.; Hirai, A.; Hirai, M.Y. A chloroplastic UDP-glucose pyrophosphorylase from Arabidopsis is the committed enzyme for the first step of sulfolipid biosynthesis. Plant Cell 2009, 21, 892–909. [Google Scholar] [CrossRef]
- Cheng, M.X.; Yuan, H.R.; Wang, R.H.; Wang, W.; Zhang, L.C.; Fan, F.F.; Li, S.Q. Identification and characterization of BES1 genes involved in grain size development of Oryza sativa L. Int. J. Biol. Macromol. 2023, 253, 127327. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Yang, D.C.; Meng, Y.Q.; Jin, J.P.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2020, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Guo, Z.H.; Ma, P.F.; Yang, G.Q.; Hu, J.Y.; Liu, Y.L.; Xia, E.H.; Zhong, M.C.; Zhao, L.; Sun, G.L.; Xu, Y.X. Genome sequences provide insights into the reticulate origin and unique traits of woody bamboos. Mol. Plant 2019, 12, 1353–1365. [Google Scholar] [CrossRef]
- Panchy, N.; Lehti Shiu, M.; Shiu, S.H. Evolution of gene duplication in plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef]
- Maere, S.; De Bodt, S.; Raes, J.; Casneuf, T.; Van Montagu, M.; Kuiper, M.; Van de Peer, Y. Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 5454–5459. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Flagel, L.E.; Paterson, A.H.; Rapp, R.A.; Soltis, D.E.; Soltis, P.S.; Wendel, J.F. Evolutionary genetics of genome merger and doubling in plants. Annu. Rev. Genet. 2008, 42, 443–461. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, D.Q.; Zhang, S.Q.; Lou, Y.F.; An, X.M.; Jiang, Z.H.; Gao, Z.M. Transcriptome and miRNAome analysis reveals components regulating tissue differentiation of bamboo shoots. Plant Physiol. 2022, 188, 2182–2198. [Google Scholar] [CrossRef]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef]
- Letunic, I.; Doerks, T.; Bork, P. SMART: Recent updates, new developments and status in 2015. Nucleic Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Almagro Armenteros, J.J.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. biorxiv 2022. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Geourjon, C.; Deleage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 1995, 11, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Tang, H.B.; DeBarry, J.D.; Tan, X.; Li, J.P.; Wang, X.Y.; Lee, T.H.; Jin, H.Z.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Wu, C.; Li, X.; Xu, J.; Cheng, Z.; Gao, J. Characterization, Expression Profile Analysis, and Functional Prediction of UGP Gene Family in Dendrocalamus brandisii. Plants 2025, 14, 1458. https://doi.org/10.3390/plants14101458
Li H, Wu C, Li X, Xu J, Cheng Z, Gao J. Characterization, Expression Profile Analysis, and Functional Prediction of UGP Gene Family in Dendrocalamus brandisii. Plants. 2025; 14(10):1458. https://doi.org/10.3390/plants14101458
Chicago/Turabian StyleLi, He, Chongyang Wu, Xiangyi Li, Junlei Xu, Zhanchao Cheng, and Jian Gao. 2025. "Characterization, Expression Profile Analysis, and Functional Prediction of UGP Gene Family in Dendrocalamus brandisii" Plants 14, no. 10: 1458. https://doi.org/10.3390/plants14101458
APA StyleLi, H., Wu, C., Li, X., Xu, J., Cheng, Z., & Gao, J. (2025). Characterization, Expression Profile Analysis, and Functional Prediction of UGP Gene Family in Dendrocalamus brandisii. Plants, 14(10), 1458. https://doi.org/10.3390/plants14101458