

DEAD-Box RNA Helicase Family in Physic Nut (Jatropha curcas L.): Structural Characterization and Response to Salinity
 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Material and Methods
2.1. Identification of Putative JcDHX Proteins and Genes
2.2. Promoter Analysis
2.3. Structural Characterization of the JcDHX Candidates and Potential Subcellular Localization
2.4. Phenetic Analysis and Orthology
2.5. Secondary Structure Elements and 3D Modeling
2.6. RNA-Seq Analysis of JcDHX Candidates and Gene Expression Validation by qPCR
2.7. Protein–Protein Interaction (PPI) Networks for JcDHX Candidates
3. Results
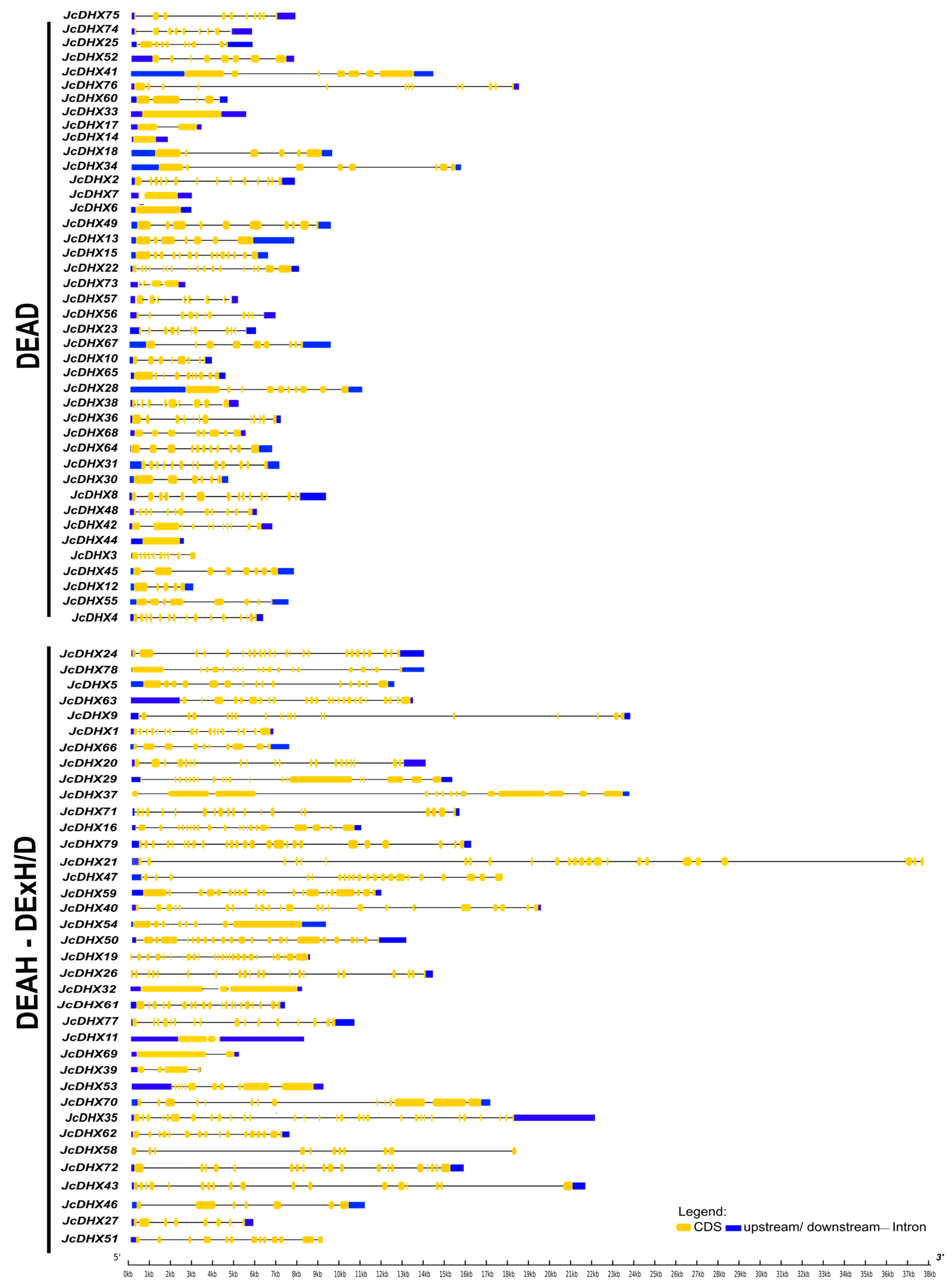
3.1. Identification of JcDHX Genes and Their Gene Structures
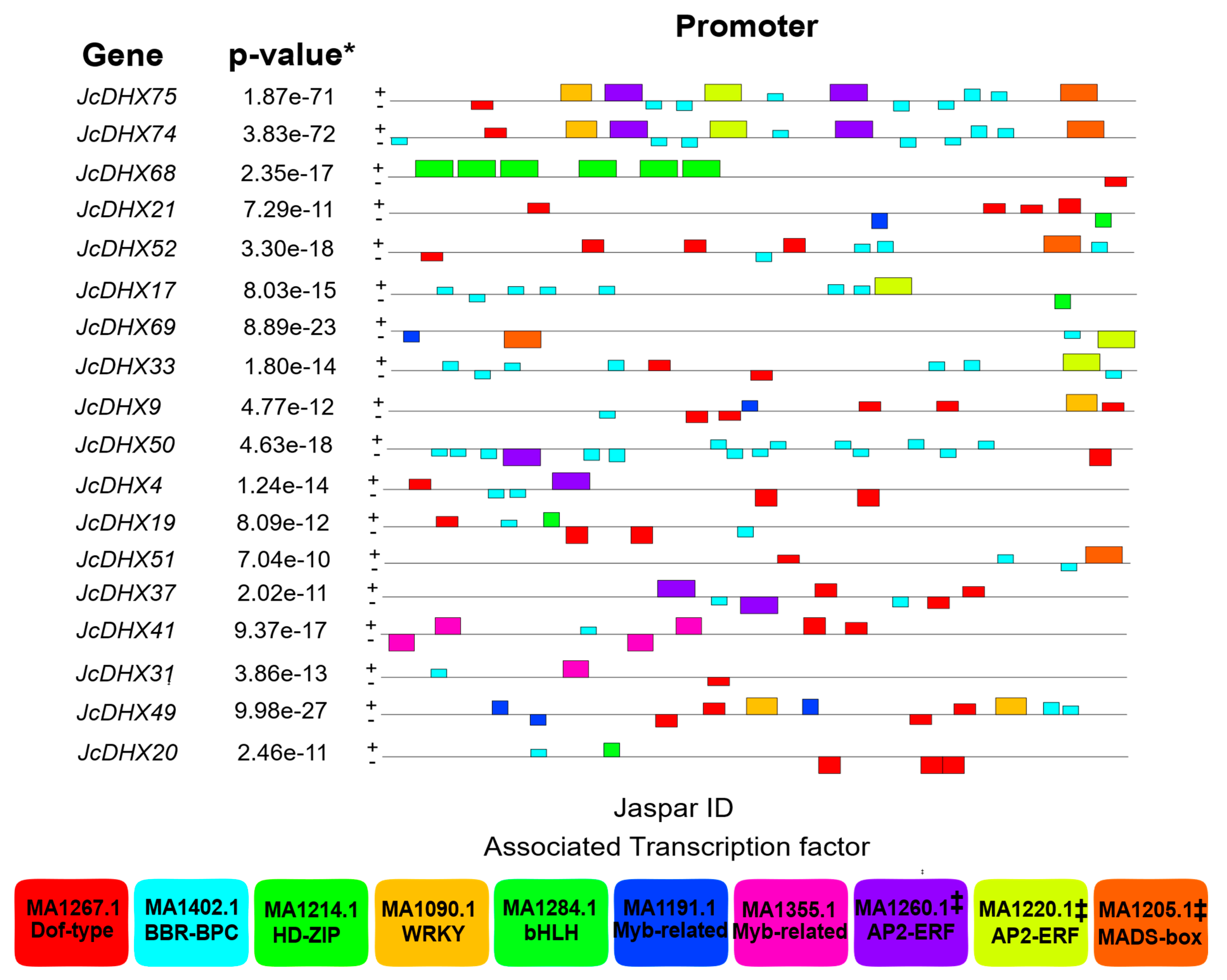
3.2. Analysis of JcDHX Gene Promoter Regions
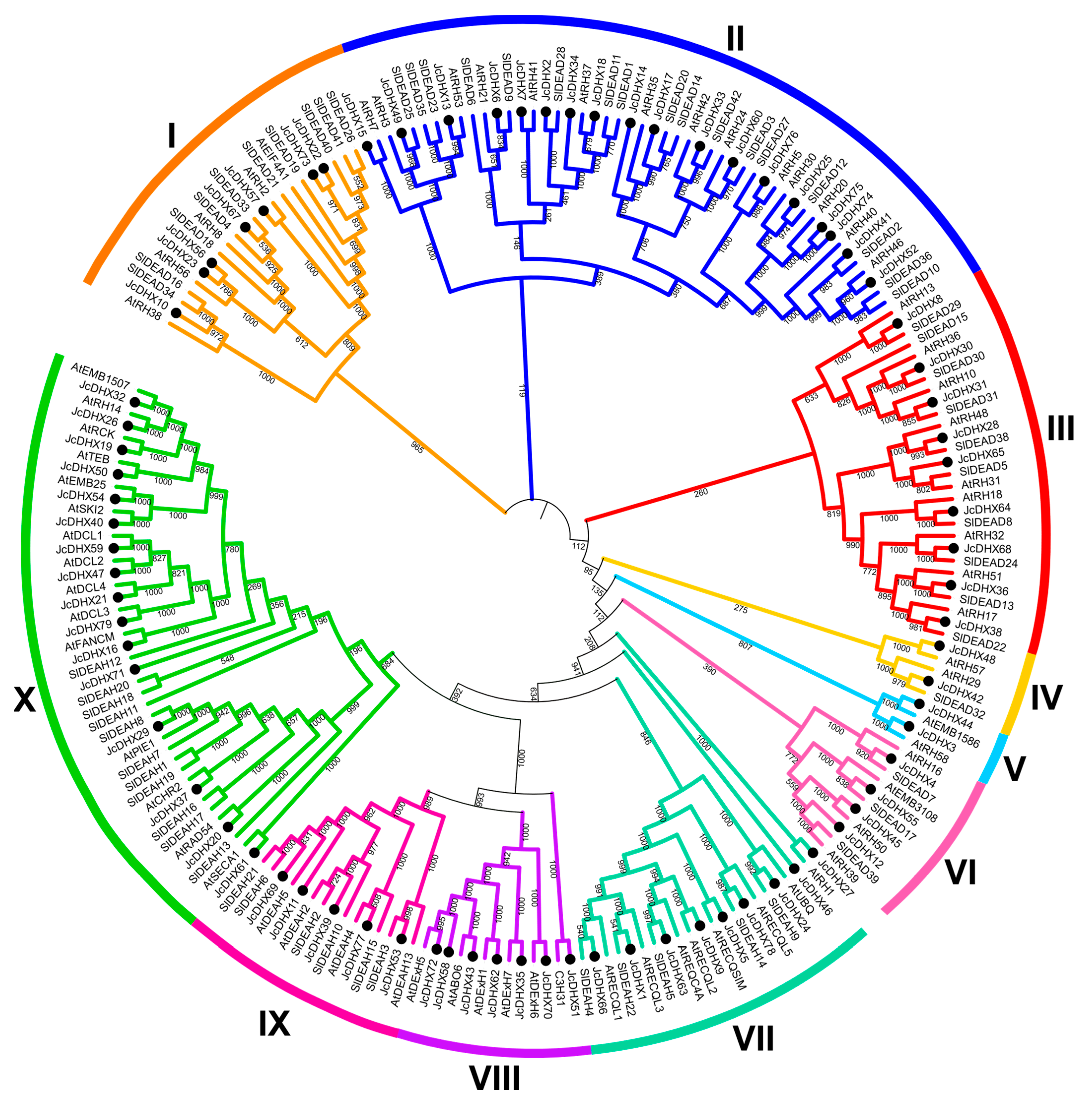
3.3. Orthology Analysis of JcDHX Genes
3.4. Phenetic Analysis of JcDHX Proteins
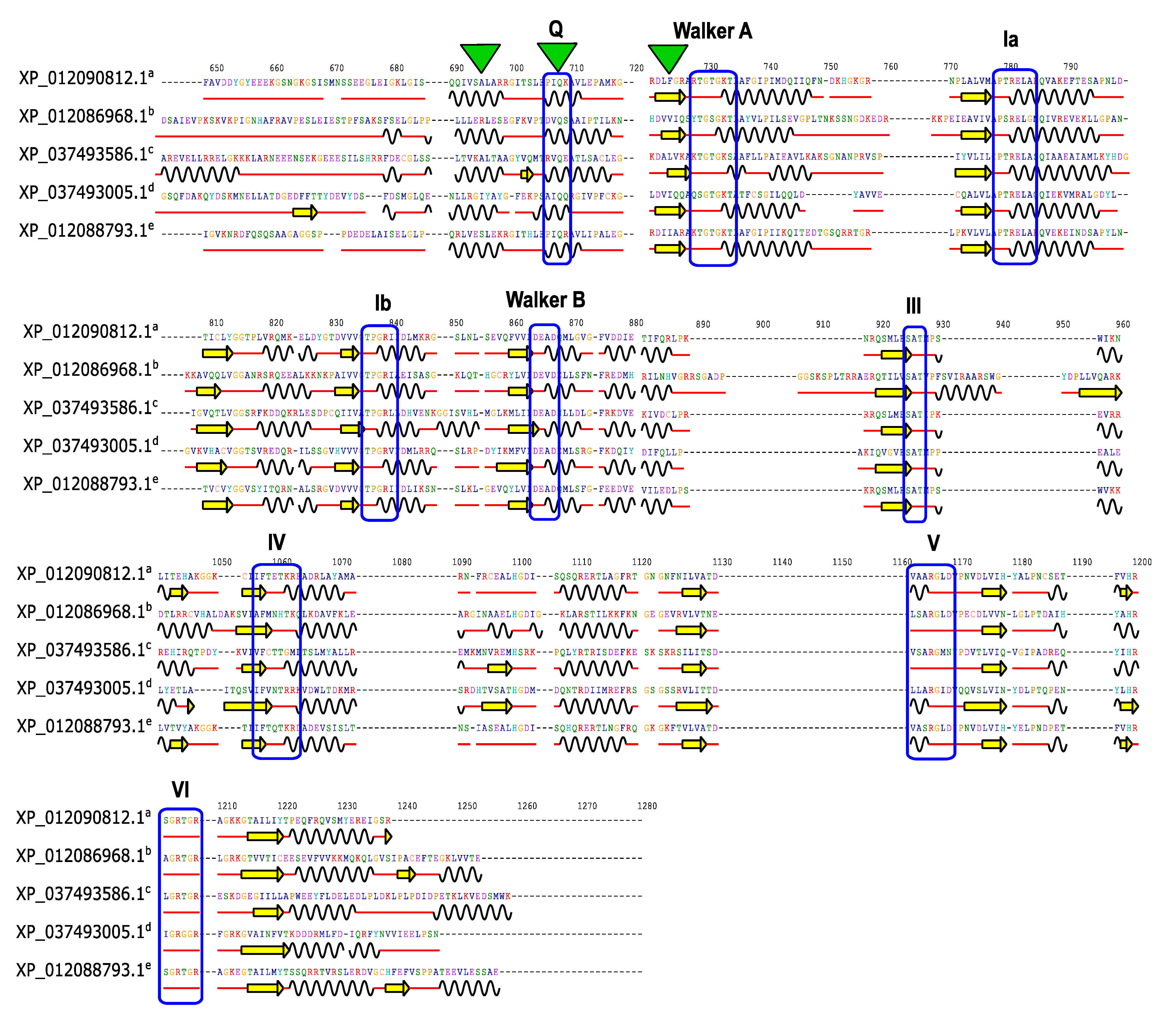
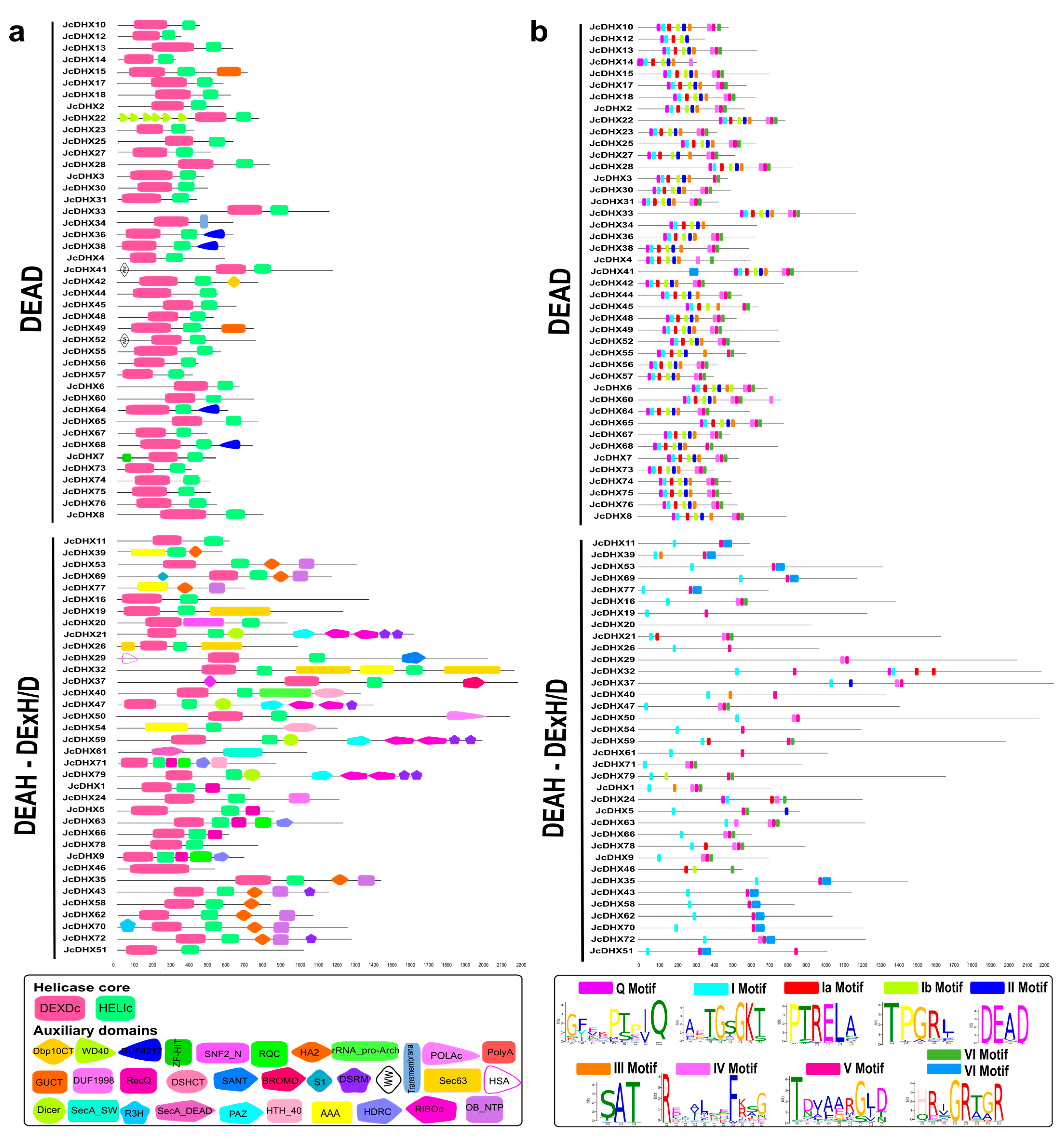
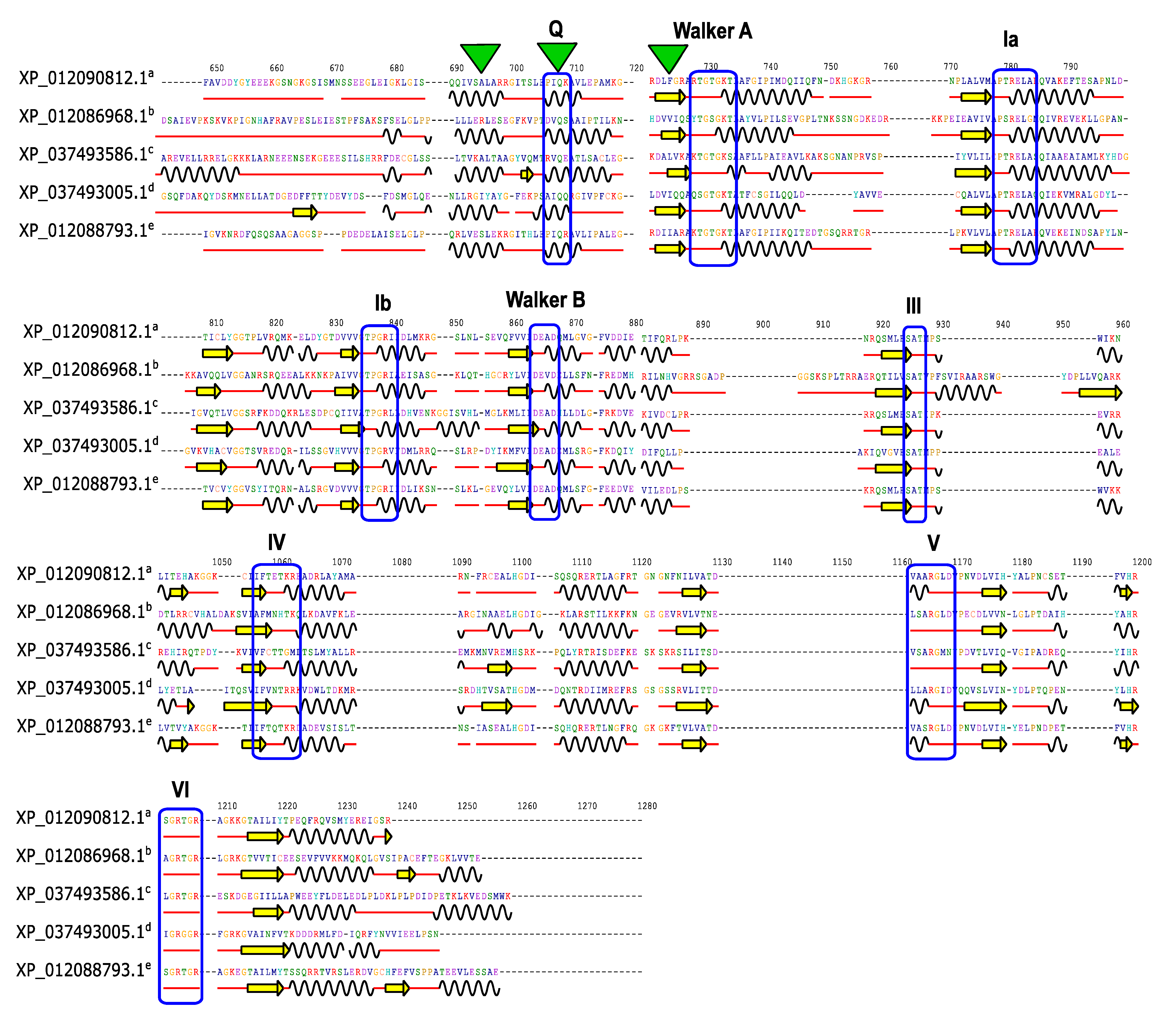
3.5. Conserved Domains and Motifs in JcDHX Proteins
3.6. Physicochemical Characteristics and Subcellular Localization of JcDHX Proteins
3.7. Prediction of Secondary Structure Elements in JcDHX Proteins
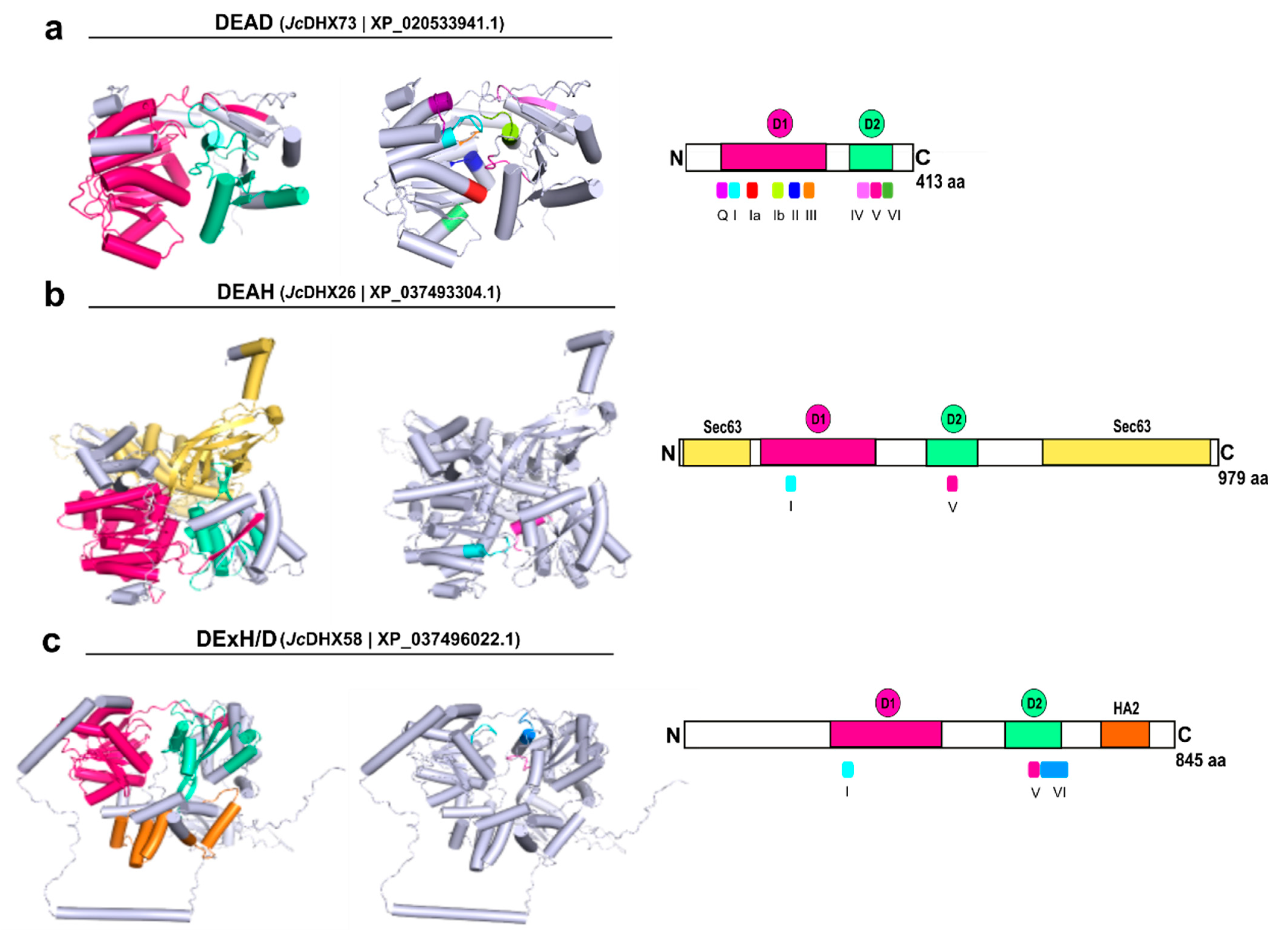
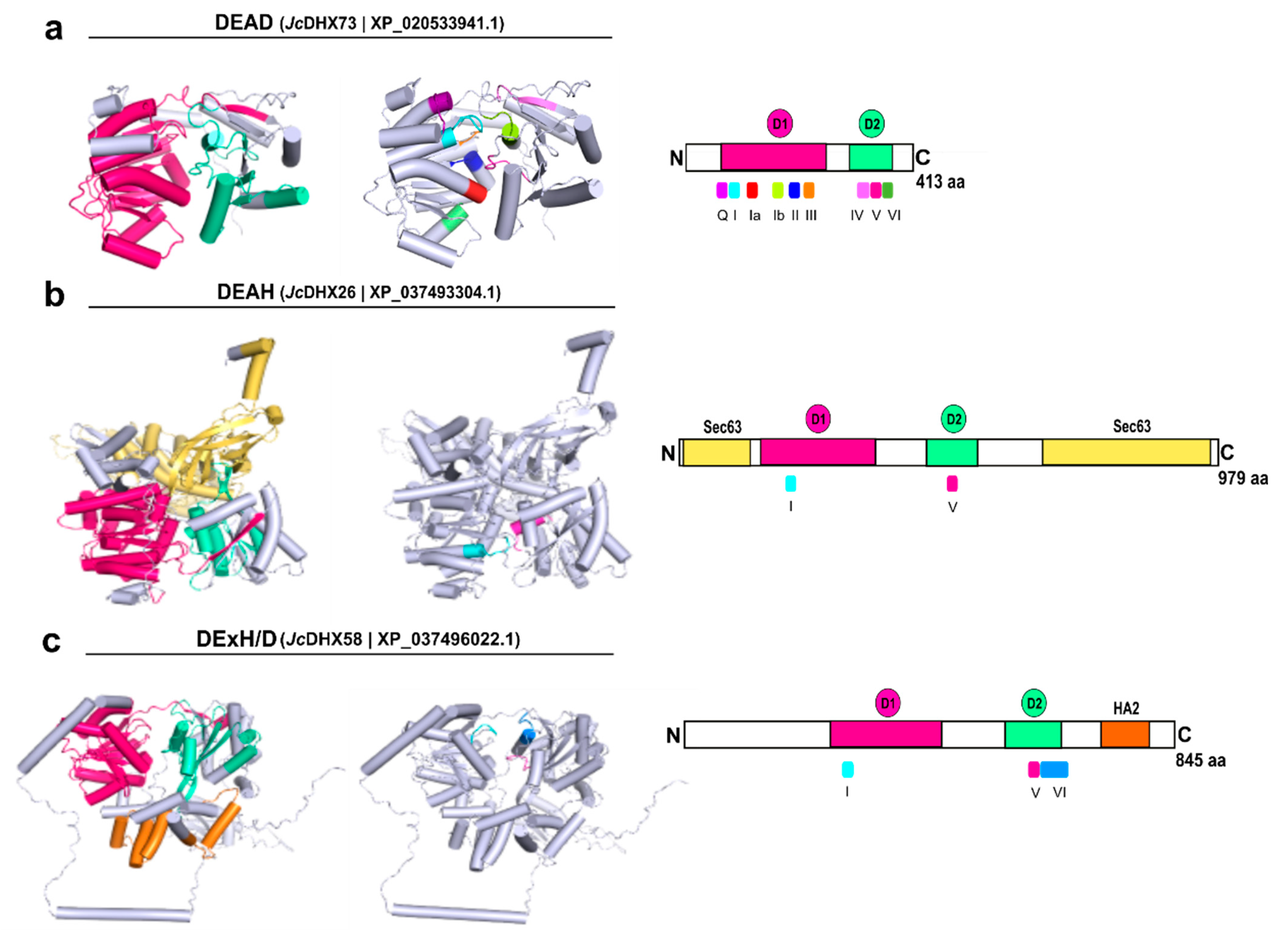
3.8. Homology Modeling of JcDHX Candidates
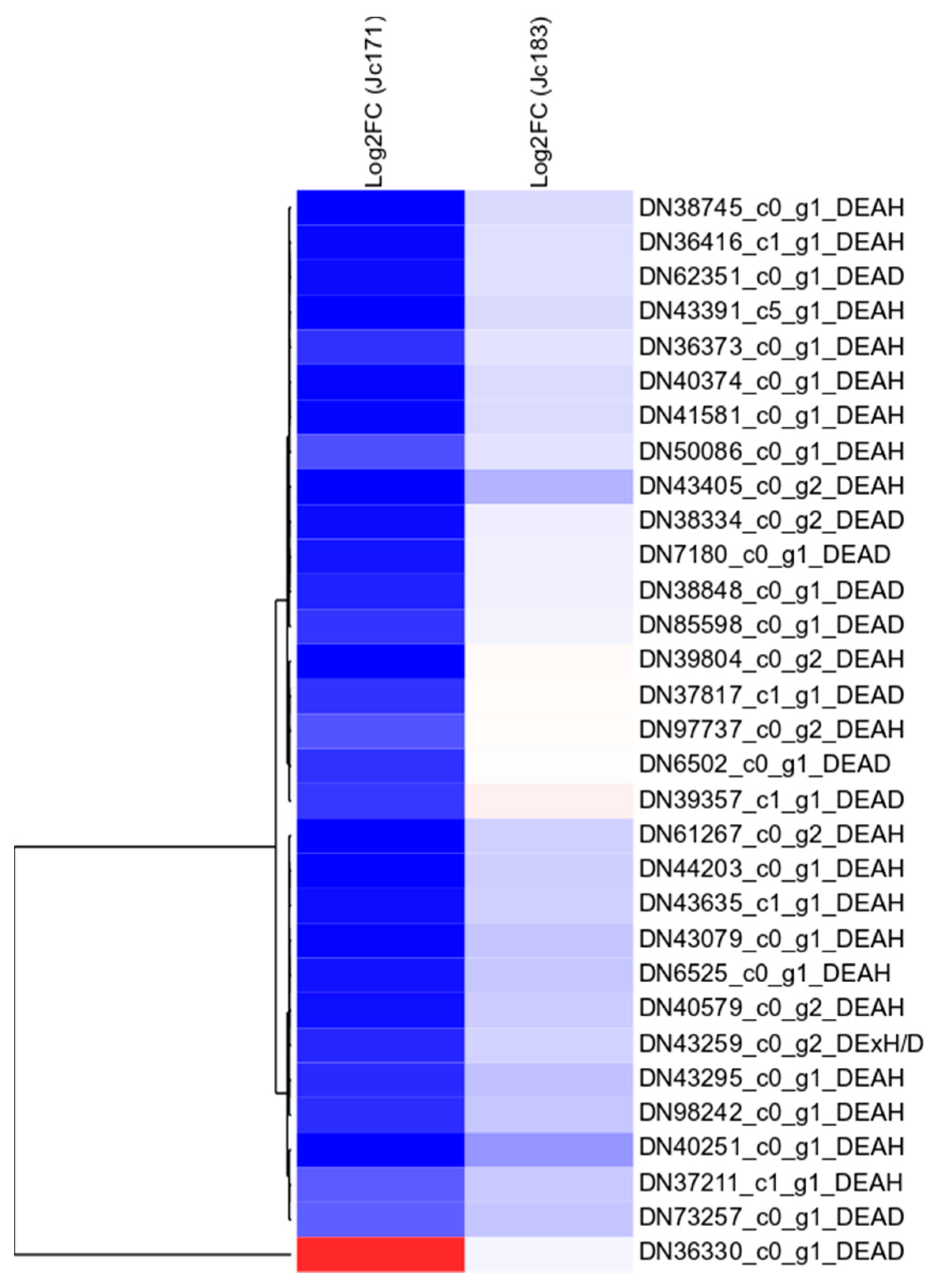
3.9. In Silico Expression of JcDHX Candidates and the qPCR Assay
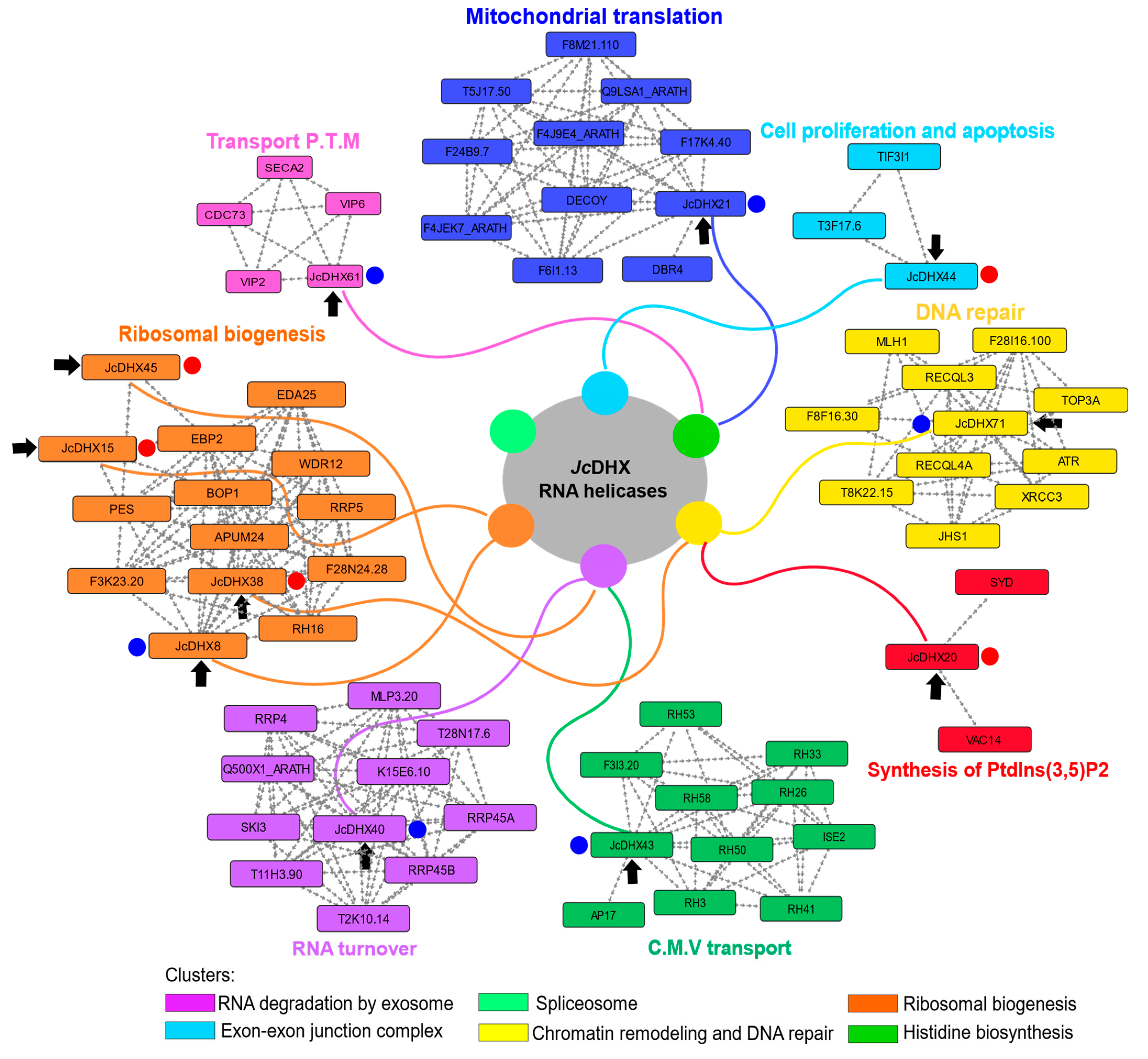
3.10. Protein–Protein Interaction Network
4. Discussion
4.1. Comprehensive Analysis of the DEAD-Box RNA Helicase Family in J. curcas Genome
4.2. Regulatory Landscape of JcDHX Genes: Insights into Cis-Regulatory Elements
4.3. Differential Regulation of JcDHX Genes in J. curcas under Salt Stress
4.4. Unraveling Functional Networks with JcDHX Proteins in Salinity Response
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Achten, W.M.J.; Verchot, L.; Franken, Y.J.; Mathijs, E.; Singh, V.P.; Aerts, R.; Muys, B. Jatropha Bio-Diesel Production and Use. Biomass Bioenergy 2008, 32, 1063–1084. [Google Scholar] [CrossRef]
- Reubens, B.; Achten, W.M.J.; Maes, W.H.; Danjon, F.; Aerts, R.; Poesen, J.; Muys, B. More than Biofuel? Jatropha curcas Root System Symmetry and Potential for Soil Erosion Control. J. Arid Environ. 2011, 75, 201–205. [Google Scholar] [CrossRef]
- Mensah, M.K.; Drebenstedt, C.; Ola, I.M.; Hoth, N.; Damptey, F.G.; Wiafe, E.D. Immobilization Effects of Co-Pyrolyzed Neem Seed Mixed with Poultry Manure on Potentially Toxic Elements in Soil and the Phytoremediation Potentials of Native Manihot Esculenta and Jatropha curcas in Ensuring Sustainable Land Use. Environ. Monit. Assess. 2023, 195, 793. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, Z.; Faizan, S.; Gulzar, B. Salt Stress, Its Impacts on Plants and the Strategies Plants Are Employing against It: A Review. J. Appl. Biol. Biotechnol. 2020, 8, 81–91. [Google Scholar] [CrossRef]
- De La Cruz, J.; Kressler, D.; Linder, P. Unwinding RNA in Saccharomyces cerevisiae: DEAD-box proteins and related families. Trends Biochem. Sci. 1999, 24, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Cordin, O.; Banroques, J.; Tanner, N.K.; Linder, P. The DEAD-Box Protein Family of RNA Helicases. Gene 2006, 367, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Umate, P.; Tuteja, R.; Tuteja, N. Genome-Wide Analysis of Helicase Gene Family from Rice and Arabidopsis: A Comparison with Yeast and Human. Plant Mol. Biol. 2010, 73, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhang, S.; Lu, L.; Cao, H.; Zheng, C. A Genome-Wide Analysis of the RNA Helicase Gene Family in Solanum Lycopersicum. Gene 2013, 513, 128–140. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Y.; Liu, J.; Xia, M.; Wang, W.; Shen, F. Genome-Wide Analysis of the RNA Helicase Gene Family in Gossypium Raimondii. Int. J. Mol. Sci. 2014, 15, 4635–4656. [Google Scholar] [CrossRef]
- Pandey, S.; Muthamilarasan, M.; Sharma, N.; Chaudhry, V.; Dulani, P.; Shweta, S.; Jha, S.; Mathur, S.; Prasad, M. Characterization of DEAD-Box Family of RNA Helicases in Tomato Provides Insights into Their Roles in Biotic and Abiotic Stresses. Environ. Exp. Bot. 2019, 158, 107–116. [Google Scholar] [CrossRef]
- Yang, S.-d.; Guo, D.-l.; Pei, M.-s.; Wei, T.-l.; Liu, H.-n.; Bian, L.; Yu, K.-k.; Zhang, G.-h.; Yu, Y.-h. Identification of the DEAD-Box RNA Helicase Family Members in Grapevine Reveals That VviDEADRH25a Confers Tolerance to Drought Stress. J. Integr. Agric. 2022, 21, 1357–1374. [Google Scholar] [CrossRef]
- Ru, J.-N.; Hou, Z.-H.; Zheng, L.; Zhao, Q.; Wang, F.-Z.; Chen, J.; Zhou, Y.-B.; Chen, M.; Ma, Y.-Z.; Xi, Y.-J.; et al. Genome-Wide Analysis of DEAD-Box RNA Helicase Family in Wheat (Triticum aestivum) and Functional Identification of TaDEAD-Box57 in Abiotic Stress Responses. Front. Plant Sci. 2021, 12, 797276. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, G.; Lee, K.; Park, S.J.; Kim, Y.O.; Kang, H. A Chloroplast-Targeted Cabbage DEAD-Box RNA Helicase BrRH22 Confers Abiotic Stress Tolerance to Transgenic Arabidopsis Plants by Affecting Translation of Chloroplast Transcripts. Plant Physiol. Biochem. 2018, 127, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Chen, G.; Dong, T.; Wang, L.; Zhang, J.; Zhao, Z.; Hu, Z. SlDEAD31, a Putative DEAD-Box RNA Helicase Gene, Regulates Salt and Drought Tolerance and Stress-Related Genes in Tomato. PLoS ONE 2015, 10, e0133849. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wan, S.; Liu, H.; Fan, S.; Zhang, Y.; Wang, W.; Xia, M.; Yuan, R.; Deng, F.; Shen, F. Over Expression of an Apocynum venetum DEAD-Box Heli Case Gene (AvDH1) in Cotton Confers Salinity Tolerance and Increases Yield in a Saline Field. Front. Plant Sci. 2016, 6, 1227. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, G.; Kang, H. Rice OsRH58, a Chloroplast DEAD-Box RNA Helicase, Improves Salt or Drought Stress Tolerance in Arabidopsis by Affecting Chloroplast Translation. BMC Plant Biol. 2019, 19, 17. [Google Scholar] [CrossRef]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2023, 51, D418–D427. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s Conserved Domain Database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent Updates, New Developments and Status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef]
- Bailey, T.; Elkan, C. Fitting a Mixture Model by Expectation Maximization; AAAI Press: Cambridge, MA, USA, 1994; pp. 3–9. [Google Scholar]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying Similarity between Motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An Upgraded Gene Feature Visualization Server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Humana Press: Totowa, NJ, USA, 2005; pp. 571–608. [Google Scholar]
- Yu, C.-S.; Chen, Y.-C.; Lu, C.-H.; Hwang, J.-K. Prediction of Protein Subcellular Localization. Proteins 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A Genomic Perspective on Protein Families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Geourjon, C.; Deleage, G. SOPMA: Significant Improvements in Protein Secondary Structure Prediction by Consensus Prediction from Multiple Alignments. Bioinformatics 1995, 11, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Alva, V.; Nam, S.Z.; Soding, J.; Lupas, A.N. The MPI Bioinformatics Toolkit as an Integrative Platform for Advanced Protein Sequence and Structure Analysis. Nucleic Acids Res. 2016, 44, W410–W415. [Google Scholar] [CrossRef] [PubMed]
- Lotun, D.P.; Cochard, C.; Vieira, F.R.J.; Bernardes, J.S. 2dSS: A Web Server for Protein Secondary Structure Visualization. bioRxiv 2019, 649426. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Lozano-Isla, F.; Campos, M.L.O.; Endres, L.; Bezerra-Neto, E.; Pompelli, M.F. Effects of Seed Storage Time and Salt Stress on the Germination of Jatropha curcas L. Ind. Crops Prod. 2018, 118, 214–224. [Google Scholar] [CrossRef]
- Souza, M.C.P.; da Silva, M.D.; Binneck, E.; de Lima Cabral, G.A.; Iseppon, A.M.B.; Pompelli, M.F.; Endres, L.; Kido, E.A. RNA-Seq Transcriptome Analysis of Jatropha curcas L. Accessions after Salt Stimulus and Unigene-Derived Microsatellite Mining. Ind. Crops Prod. 2020, 147, 112168. [Google Scholar] [CrossRef]
- Haas, B.; Papanicolaou, A.; Yassour, M. TransDecoder. 2017. Available online: https://github.com/TransDecoder/TransDecoder (accessed on 17 August 2021).
- Camargo, A.P.; Sourkov, V.; Pereira, G.A.G.; Carazzolle, M.F. RNAsamba: Neural Network-Based Assessment of the Protein-Coding Potential of RNA Sequences. NAR Genom. Bioinform. 2020, 2, lqz024. [Google Scholar] [CrossRef]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bairoch, A. UniProtKB/Swiss-Prot: The Manually Annotated Section of the UniProt KnowledgeBase. In Plant Bioinformatics: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2007; pp. 89–112. [Google Scholar]
- Wheeler, D.L.; Church, D.M.; Federhen, S.; Lash, A.E.; Madden, T.L.; Pontius, J.U.; Schuler, G.D.; Schriml, L.M.; Sequeira, E.; Tatusova, T.A. Database Resources of the National Center for Biotechnology. Nucleic Acids Res. 2003, 31, 28–33. [Google Scholar] [CrossRef]
- Saldanha, A.J. Java Treeview—Extensible Visualization of Microarray Data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef] [PubMed]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for General Users and for Biologist Programmers. Methods Mol. Biol. 2000, 132, 365–386. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, R. Quantification on the LightCycler. In Rapid Cycle Real-Time PCR: Methods and Applications; Springer: Berlin/Heidelberg, Germany, 2001; pp. 21–34. [Google Scholar]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative Expression Software Tool (REST) for Group-Wise Comparison and Statistical Analysis of Relative Expression Results in Real-Time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S. The STRING Database in 2023: Protein–Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Linder, P.; Lasko, P.F.; Ashburner, M.; Leroy, P.; Nielsen, P.J.; Nishi, K.; Schnier, J.; Slonimski, P.P. Birth of the DEAD Box. Nature 1989, 337, 121–122. [Google Scholar] [CrossRef] [PubMed]
- Aubourg, S.; Kreis, M.; Lecharny, A. The DEAD Box RNA Helicase Family in Arabidopsis Thaliana. Nucleic Acids Res. 1999, 27, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhang, S.; Huang, J.; Zheng, C. Genome-Wide Comparative in Silico Analysis of the RNA Helicase Gene Family in Zea Mays and Glycine Max: A Comparison with Arabidopsis and Oryza Sativa. PLoS ONE 2013, 8, e78982. [Google Scholar] [CrossRef] [PubMed]
- Jo, B.-S.; Choi, S.S. Introns: The Functional Benefits of Introns in Genomes. Genom. Inform. 2015, 13, 112. [Google Scholar] [CrossRef]
- Kammel, C.; Thomaier, M.; Sørensen, B.B.; Schubert, T.; Längst, G.; Grasser, M.; Grasser, K.D. Arabidopsis DEAD-Box RNA Helicase UAP56 Interacts with Both RNA and DNA as Well as with MRNA Export Factors. PLoS ONE 2013, 8, e0060644. [Google Scholar] [CrossRef]
- Huang, C.K.; Shen, Y.L.; Huang, L.F.; Wu, S.J.; Yeh, C.H.; Lu, C.A. The DEAD-Box RNA Helicase AtRH7/PRH75 Participates in Pre-RRNA Processing, Plant Development and Cold Tolerance in Arabidopsis. Plant Cell Physiol. 2016, 57, 174–191. [Google Scholar] [CrossRef] [PubMed]
- Lange, H.; Zuber, H.; Sement, F.M.; Chicher, J.; Kuhn, L.; Hammann, P.; Brunaud, V.; Bérard, C.; Bouteiller, N.; Balzergue, S.; et al. The RNA Helicases AtMTR4 and HEN2 Target Specific Subsets of Nuclear Transcripts for Degradation by the Nuclear Exosome in Arabidopsis Thaliana. PLoS Genet. 2014, 10, e1004564. [Google Scholar] [CrossRef]
- Nawaz, G.; Kang, H. Chloroplast- or Mitochondria-Targeted DEAD-Box RNA Helicases Play Essential Roles in Organellar RNA Metabolism and Abiotic Stress Responses. Front. Plant Sci. 2017, 8, 871. [Google Scholar] [CrossRef]
- Jermy, A.J.; Willer, M.; Davis, E.; Wilkinson, B.M.; Stirling, C.J. The Brl Domain in Sec63p Is Required for Assembly of Functional Endoplasmic Reticulum Translocons. J. Biol. Chem. 2006, 281, 7899–7906. [Google Scholar] [CrossRef]
- Cordin, O.; Tanner, N.K.; Doere, M.; Linder, P.; Banroques, J. The Newly Discovered Q Motif of DEAD-box RNA Helicases Regulates RNA-binding and Helicase Activity. EMBO J. 2004, 23, 2478–2487. [Google Scholar] [CrossRef]
- Rogers, G.W., Jr.; Komar, A.A.; Merrick, W.C. EIF4A: The Godfather of the DEAD Box Helicases. Prog. Nucleic Acid Res. Mol. Biol. 2002, 72, 307–331. [Google Scholar]
- Story, R.M.; Li, H.; Abelson, J.N. Crystal Structure of a DEAD Box Protein from the Hyperthermophile Methanococcus Jannaschii. Proc. Natl. Acad. Sci. USA 2001, 98, 1465–1470. [Google Scholar] [CrossRef]
- Tanner, N.K.; Linder, P. DExD/H Box RNA Helicases: From Generic Motors to Specific Dissociation Functions. Mol. Cell 2001, 8, 251–262. [Google Scholar] [CrossRef]
- Caruthers, J.M.; McKay, D.B. Helicase Structure and Mechanism. Curr. Opin. Struct. Biol. 2002, 12, 123–133. [Google Scholar] [CrossRef]
- Fairman-Williams, M.E.; Guenther, U.-P.; Jankowsky, E. SF1 and SF2 Helicases: Family Matters. Curr. Opin. Struct. Biol. 2010, 20, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Nolan, T.M.; Jiang, H.; Yin, Y. AP2/ERF Transcription Factor Regulatory Networks in Hormone and Abiotic Stress Responses in Arabidopsis. Front. Plant Sci. 2019, 10, 228. [Google Scholar] [CrossRef]
- Li, C.; Yan, C.; Sun, Q.; Wang, J.; Yuan, C.; Mou, Y.; Shan, S.; Zhao, X. The BHLH Transcription Factor AhbHLH112 Improves the Drought Tolerance of Peanut. BMC Plant Biol. 2021, 21, 540. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, Z.; Zhang, Y.; Guo, J.; Liu, L.; Wang, C.; Wang, B.; Han, G. The Roles of HD-ZIP Proteins in Plant Abiotic Stress Tolerance. Front. Plant Sci. 2022, 13, 1027071. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Wang, S.; Zhang, T.; Xu, L.; Li, Y.; Chao, Y.; Han, L. Expression of the Medicago Truncatula MtDof32 Transcription Factor Regulates Plant Growth and Enhances Abiotic Stress Tolerances in Transgenic Arabidopsis. Environ. Exp. Bot. 2021, 183, 104339. [Google Scholar] [CrossRef]
- Su, Y.; Liang, W.; Liu, Z.; Wang, Y.; Zhao, Y.; Ijaz, B.; Hua, J. Overexpression of GhDof1 Improved Salt and Cold Tolerance and Seed Oil Content in Gossypium Hirsutum. J. Plant Physiol. 2017, 218, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tian, M.; Feng, Z.; Zhang, J.; Lu, J.; Fu, X.; Ma, L.; Wei, H.; Wang, H. GhDof1. 7, a Dof Transcription Factor, Plays Positive Regulatory Role under Salinity Stress in Upland Cotton. Plants 2023, 12, 3740. [Google Scholar] [CrossRef]
- Bakshi, R.; Prakash, T.; Dash, D.; Brahmachari, V. In Silico Characterization of the INO80 Subfamily of SWI2/SNF2 Chromatin Remodeling Proteins. Biochem. Biophys. Res. Commun. 2004, 320, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Sun, Y.; Ahmed, R.I.; Ren, A.; Xie, M. Research Progress on Plant RING-Finger Proteins. Genes 2019, 10, 973. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.; Fraser, C.S.; Marunde, M.R.; Parker, M.M.; Sagum, C.; Burg, J.M.; Hall, N.; Popova, I.K.; Rodriguez, K.L.; Vaidya, A. Characterization of the Plant Homeodomain (PHD) Reader Family for Their Histone Tail Interactions. Epigenet. Chromatin 2020, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Babu, M.; Aravind, L. The HIRAN Domain and Recruitment of Chromatin Remodeling and Repair Activities to Damaged DNA. Cell Cycle 2006, 5, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhong, X.; Bernatavichute, Y.V.; Stroud, H.; Feng, S.; Caro, E.; Vashisht, A.A.; Terragni, J.; Chin, H.G.; Tu, A. Dual Binding of Chromomethylase Domains to H3K9me2-Containing Nucleosomes Directs DNA Methylation in Plants. Cell 2012, 151, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Qian, B.; Cao, F.; Wu, W.; Yang, L.; Guan, Q.; Gu, X.; Wang, P.; Okusolubo, T.A.; Dunn, S.L. An Arabidopsis PWI and RRM Motif-Containing Protein Is Critical for Pre-MRNA Splicing and ABA Responses. Nat. Commun. 2015, 6, 8139. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Cai, Q.; Qiao, L.; Huang, C.-Y.; Wang, S.; Miao, W.; Ha, T.; Wang, Y.; Jin, H. RNA-Binding Proteins Contribute to Small RNA Loading in Plant Extracellular Vesicles. Nat. Plants 2021, 7, 342–352. [Google Scholar] [CrossRef]
- Fukudome, A.; Fukuhara, T. Plant Dicer-like Proteins: Double-Stranded RNA-Cleaving Enzymes for Small RNA Biogenesis. J. Plant Res. 2017, 130, 33–44. [Google Scholar] [CrossRef]
- Cai, J.; Meng, X.; Li, G.; Dong, T.; Sun, J.; Xu, T.; Li, Z.; Han, Y.; Zhu, M. Identification, Expression Analysis, and Function Evaluation of 42 Tomato DEAD-Box RNA Helicase Genes in Growth Development and Stress Response. Acta Physiol. Plant. 2018, 40, 94. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, K.A.; Oh, T.R.; Park, C.M.; Kang, H. Functional Characterization of DEAD-Box RNA Helicases in Arabidopsis Thaliana under Abiotic Stress Conditions. Plant Cell Physiol. 2008, 49, 1563–1571. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Seok, H.Y.; Woo, D.H.; Lee, S.Y.; Moon, Y.H. Overexpression of the DEAD-Box RNA Helicase Gene AtRH17 Confers Tolerance to Salt Stress in Arabidopsis. Int. J. Mol. Sci. 2018, 19, 3777. [Google Scholar] [CrossRef] [PubMed]
- Banu, M.S.A.; Huda, K.M.K.; Harun-Ur-Rashid, M.; Parveen, S.; Tuteja, N. A DEAD Box Helicase Psp68 Positively Regulates Salt Stress Responses in Marker-Free Transgenic Rice Plants. Transgenic Res. 2023, 32, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Gu, H.; Jiang, W.; Tian, Z.; Shi, G.; Chen, W.; Tian, B.; Wei, X.; Zhang, L.; Wei, F.; et al. BrDHC1, a Novel Putative DEAD-Box Helicase Gene, Confers Drought Tolerance in Transgenic Brassica Rapa. Horticulturae 2022, 8, 707. [Google Scholar] [CrossRef]
- Kant, P.; Kant, S.; Gordon, M.; Shaked, R.; Barak, S. STRESS RESPONSE SUPPRESSOR1 and STRESS RESPONSE SUPPRESSOR2, Two DEAD-Box RNA Helicases That Attenuate Arabidopsis Responses to Multiple Abiotic Stresses. Plant Physiol. 2007, 145, 814–830. [Google Scholar] [CrossRef]
- de Lima Cabral, G.A.; Binneck, E.; de Souza, M.C.P.; da Silva, M.D.; Costa Ferreira Neto, J.R.; Pompelli, M.F.; Endres, L.; Kido, É.A. First Expressed TFome of Physic Nut (Jatropha curcas L.) After Salt Stimulus. Plant Mol. Biol. Report. 2020, 38, 189–208. [Google Scholar] [CrossRef]
- Silva-Santos, L.; Corte-Real, N.; Dias-Pereira, J.; Figueiredo, R.C.B.Q.; Endres, L.; Pompelli, M.F. Salinity Shock in Jatropha curcas Leaves Is More Pronounced during Recovery than during Stress Time. bioRxiv 2018, 378208. [Google Scholar] [CrossRef]
- Ding, F.; Cui, P.; Wang, Z.; Zhang, S.; Ali, S.; Xiong, L. Genome-Wide Analysis of Alternative Splicing of Pre-MRNA under Salt Stress in Arabidopsis. BMC Genom. 2014, 15, 431. [Google Scholar] [CrossRef]
- Zhu, G.; Li, W.; Zhang, F.; Guo, W. RNA-Seq Analysis Reveals Alternative Splicing under Salt Stress in Cotton, Gossypium davidsonii. BMC Genom. 2018, 19, 73. [Google Scholar] [CrossRef]
- Wang, B.; Chai, H.; Zhong, Y.; Shen, Y.; Yang, W.; Chen, J.; Xin, Z.; Shi, H. The DEAD-Box RNA Helicase SHI2 Functions in Repression of Salt-Inducible Genes and Regulation of Cold-Inducible Gene Splicing. J. Exp. Bot. 2020, 71, 1598–1613. [Google Scholar] [CrossRef]
- Ji, H.; Yang, G.; Zhang, X.; Zhong, Q.; Qi, Y.; Wu, K.; Shen, T. Regulation of Salt Tolerance in the Roots of Zea Mays by L-Histidine through Transcriptome Analysis. Front. Plant Sci. 2022, 13, 1049954. [Google Scholar] [CrossRef]
- Lin, H.H.; Lin, K.H.; Syu, J.Y.; Tang, S.Y.; Lo, H.F. Physiological and Proteomic Analysis in Two Wild Tomato Lines under Waterlogging and High Temperature Stress. J. Plant Biochem. Biotechnol. 2016, 25, 87–96. [Google Scholar] [CrossRef]
- Prajapati, P.; Gupta, P.; Kharwar, R.N.; Seth, C.S. Nitric Oxide Mediated Regulation of Ascorbate-Glutathione Pathway Alleviates Mitotic Aberrations and DNA Damage in Allium cepa L. under Salinity Stress. Int. J. Phytoremediat. 2023, 25, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Zhang, F. Cell Cycle Regulation in the Plant Response to Stress. Front. Plant Sci. 2020, 10, 1765. [Google Scholar] [CrossRef] [PubMed]
- Chirinos-Arias, M.C.; Spampinato, C.P. Role of the Mismatch Repair Protein MSH7 in Arabidopsis Adaptation to Acute Salt Stress. Plant Physiol. Biochem. 2021, 169, 280–290. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Regulation of DNA Repair throughout the Cell Cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Chantarachot, T.; Bailey-Serres, J. Polysomes, Stress Granules, and Processing Bodies: A Dynamic Triumvirate Controlling Cytoplasmic MRNA Fate and Function. Plant Physiol. 2018, 176, 254–269. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, S.; Shi, H.; Ma, J.; Jing, M.; Han, Y. The TSN1 Binding Protein RH31 Is a Component of Stress Granules and Participates in Regulation of Salt-Stress Tolerance in Arabidopsis. Front. Plant Sci. 2021, 12, 804356. [Google Scholar] [CrossRef]
- Kearly, A.; Nelson, A.D.L.; Skirycz, A.; Chodasiewicz, M. Composition and Function of Stress Granules and P-Bodies in Plants. Semin. Cell Dev. Biol. 2024, 156, 167–175. [Google Scholar] [CrossRef]
- Liu, Y.; Imai, R. Function of Plant DExD/H-Box RNA Helicases Associated with Ribosomal RNA Biogenesis. Front. Plant Sci. 2018, 9, 125. [Google Scholar] [CrossRef]
- Panse, V.G.; Johnson, A.W. Maturation of Eukaryotic Ribosomes: Acquisition of Functionality. Trends Biochem. Sci. 2010, 35, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, N.O.; Makarova, S.; Makhotenko, A.; Love, A.J.; Taliansky, M. The Multiple Functions of the Nucleolus in Plant Development, Disease and Stress Responses. Front. Plant Sci. 2018, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, Y.; Li, J.; Jiang, A.; Cheng, Y.; Zhang, W. Mitochondrial Proteome during Salt Stress-Induced Programmed Cell Death in Rice. Plant Physiol. Biochem. 2009, 47, 407–415. [Google Scholar] [CrossRef]
- Wang, W.; Xu, M.; Liu, X.; Tu, J. The Rice Eukaryotic Translation Initiation Factor 3 Subunit e (OseiF3e) Influences Organ Size and Pollen Maturation. Front. Plant Sci. 2016, 7, 1399. [Google Scholar] [CrossRef] [PubMed]
- Valandro, F.; Menguer, P.K.; Cabreira-Cagliari, C.; Margis-Pinheiro, M.; Cagliari, A. Programmed Cell Death (PCD) Control in Plants: New Insights from the Arabidopsis Thaliana Deathosome. Plant Sci. 2020, 299, 110603. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, X.; Liu, Y.; Zhao, X. Salt Stress Induces Programmed Cell Death in Thellungiella Halophila Suspension-Cultured Cells. J. Plant Physiol. 2010, 167, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; De Clercq, I.; Van Aken, O.; Law, S.R.; Ivanova, A.; Willems, P.; Giraud, E.; Van Breusegem, F.; Whelan, J. Anterograde and Retrograde Regulation of Nuclear Genes Encoding Mitochondrial Proteins during Growth, Development, and Stress. Mol. Plant 2014, 7, 1075–1093. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Auwerx, J. Systems Phytohormone Responses to Mitochondrial Proteotoxic Stress. Mol. Cell 2017, 68, 540–551.e5. [Google Scholar] [CrossRef]
- Depaepe, T.; Hendrix, S.; van Rensburg, H.C.J.; Van den Ende, W.; Cuypers, A.; Van Der Straeten, D. At the Crossroads of Survival and Death: The Reactive Oxygen Species–Ethylene–Sugar Triad and the Unfolded Protein Response. Trends Plant Sci. 2021, 26, 338–351. [Google Scholar] [CrossRef]
- Zou, Y.; Liu, Z.; Bai, J.; Zhou, Y.; Lu, D. Mitochondrial Proteotoxic Stresses Activate Abscisic Acid Signaling in Plants. Environ. Exp. Bot. 2023, 205, 105134. [Google Scholar] [CrossRef]
- Tuteja, N.; Sahoo, R.K.; Garg, B.; Tuteja, R. OsSUV3 Dual Helicase Functions in Salinity Stress Tolerance by Maintaining Photosynthesis and Antioxidant Machinery in Rice (Oryza sativa L. Cv. IR64). Plant J. 2013, 76, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Méndez, M.F.; Vera-Estrella, R.; Amezcua-Romero, J.C.; Rosas-Santiago, P.; Hernández-Domínguez, E.E.; de Luna-Valdez, L.A.; Pantoja, O. Mesembryanthemum crystallinum Plasma Membrane Root Aquaporins Are Regulated via Clathrin-Coated Vesicles in Response to Salt Stress. bioRxiv 2022. [Google Scholar] [CrossRef]
- Larson, E.R.; van Zelm, E.; Roux, C.; Marion-Poll, A.; Blatt, M.R. Clathrin Heavy Chain Subunits Coordinate Endo- and Exocytic Traffic and Affect Stomatal Movement. Plant Physiol. 2017, 175, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hanh Nguyen, H.; Park, Y.; Lin, J.; Hwang, I. Spatial Regulation of RBOHD via AtECA4-Mediated Recycling and Clathrin-Mediated Endocytosis Contributes to ROS Accumulation during Salt Stress Response but Not Flg22-Induced Immune Response. Plant J. 2022, 109, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Meijer, H.J.G.; Divecha, N.; Van Den Ende, H.; Musgrave, A.; Munnik, T. Hyperosmotic Stress Induces Rapid Synthesis of Phosphatidyl-d-Inositol 3,5-Bisphosphate in Plant Cells. Planta 1999, 208, 294–298. [Google Scholar] [CrossRef]
- Compton, L.M.; Ikonomov, O.C.; Sbrissa, D.; Garg, P.; Shisheva, A. Active Vacuolar H+ Atpase and Functional Cycle of Rab5 Are Required for the Vacuolation Defect Triggered by Ptdins(3,5)P2 Loss under PIKfyve or Vps34 Deficiency. Am. J. Physiol.—Cell Physiol. 2016, 311, C366–C377. [Google Scholar] [CrossRef] [PubMed]
- Bak, G.; Lee, E.J.; Lee, Y.; Kato, M.; Segami, S.; Sze, H.; Maeshima, M.; Hwang, J.U.; Lee, Y. Rapid Structural Changes and Acidification of Guard Cell Vacuoles during Stomatal Closure Require Phosphatidylinositol 3,5-Bisphosphate. Plant Cell 2013, 25, 2202–2216. [Google Scholar] [CrossRef]
- Pérez Koldenkova, V.; Hatsugai, N. Vacuolar Convolution: Possible Mechanisms and Role of Phosphatidylinositol 3,5-Bisphosphate. Funct. Plant Biol. 2017, 44, 751–760. [Google Scholar] [CrossRef]
- Li, S.C.; Diakov, T.T.; Xu, T.; Tarsio, M.; Zhu, W.; Couoh-Cardel, S.; Weisman, L.S.; Kane, P.M. The Signaling Lipid PI(3,5)P2 Stabilizes V1-Vo Sector Interactions and Activates the V-ATPase. Mol. Biol. Cell 2014, 25, 1251–1262. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Result | ||||||||
|---|---|---|---|---|---|---|---|---|
| RNA-Seq Transcript | Gene | Efficiency (%) | Relative Expression | Std. Error | 95% C.I. | P(H1) | In Silico | qPCR |
| DN43295_c0_g1_i2 | JcDHX43 | 102.34 | −0.54 | 0.237–1.111 | 0.105–1.999 | 0.047 | DR | DR |
| DN43259_c0_g2_i2 | JcDHX43 | 95.76 | −0.22 | 0.083–0.694 | 0.033–2.514 | 0.000 | DR | DR |
| DN39804_c0_g2_i2 | JcDHX8 | 108.19 | −0.30 | 0.051–1.709 | 0.024–2.832 | 0.043 | DR | DR |
| DN43635_c1_g1_i2 | JcDHX71 | 99.72 | −0.13 | 0.012–1.999 | 0.001–3.936 | 0.032 | DR | DR |
| DN36330_c0_g1_i1 | JcDHX44 | 102.98 | 3.50 | 1.451–14.283 | 1.043–30.645 | 0.000 | UR | UR |
| DN97737_c0_g2_i1 | JcDHX40 | 101.48 | −0.49 | 0.346–0.686 | 0.236–0.840 | 0.000 | DR | DR |
| DN43391_c5_g1_i3 | JcDHX40 | 97.43 | −0.51 | 0.380–0.704 | 0.296–0.867 | 0.000 | DR | DR |
| DN39804_c0_g2_i1 | JcDHX21 | 90.38 | −0.60 | 0.345–1.059 | 0.214–1.426 | 0.014 | DR | DR |
| DN41581_c0_g1_i2 | JcDHX61 | 108.16 | −0.68 | 0.398–1.046 | 0.282–2.043 | 0.031 | DR | DR |
| DN40374_c0_g1_i2 | JcDHX20 | 109.42 | 1.97 | 1.135–3.584 | 0.677–5.953 | 0.004 | DR | UR |
| DN62351_c0_g1_i1 | JcDHX38 | 95.05 | 1.78 | 1.273–2.527 | 0.785–3.596 | 0.000 | DR | UR |
| DN7180_c0_g1_i1 | JcDHX45 | 91.69 | 5.56 | 0.966–28.716 | 0.185–79.341 | 0.012 | DR | UR |
| DN85598_c0_g1_i1 | JcDHX15 | 98.48 | 1.42 | 0.942–2.299 | 0.573–3.147 | 0.042 | DR | UR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva, R.H.; Silva, M.D.d.; Ferreira-Neto, J.R.C.; Souza, B.d.B.; de Araújo, F.N.; Oliveira, E.J.d.S.; Benko-Iseppon, A.M.; da Costa, A.F.; Kido, É.A. DEAD-Box RNA Helicase Family in Physic Nut (Jatropha curcas L.): Structural Characterization and Response to Salinity. Plants 2024, 13, 905. https://doi.org/10.3390/plants13060905
da Silva RH, Silva MDd, Ferreira-Neto JRC, Souza BdB, de Araújo FN, Oliveira EJdS, Benko-Iseppon AM, da Costa AF, Kido ÉA. DEAD-Box RNA Helicase Family in Physic Nut (Jatropha curcas L.): Structural Characterization and Response to Salinity. Plants. 2024; 13(6):905. https://doi.org/10.3390/plants13060905
Chicago/Turabian Styleda Silva, Rahisa Helena, Manassés Daniel da Silva, José Ribamar Costa Ferreira-Neto, Bruna de Brito Souza, Francielly Negreiros de Araújo, Elvia Jéssica da Silva Oliveira, Ana Maria Benko-Iseppon, Antonio Félix da Costa, and Éderson Akio Kido. 2024. "DEAD-Box RNA Helicase Family in Physic Nut (Jatropha curcas L.): Structural Characterization and Response to Salinity" Plants 13, no. 6: 905. https://doi.org/10.3390/plants13060905
APA Styleda Silva, R. H., Silva, M. D. d., Ferreira-Neto, J. R. C., Souza, B. d. B., de Araújo, F. N., Oliveira, E. J. d. S., Benko-Iseppon, A. M., da Costa, A. F., & Kido, É. A. (2024). DEAD-Box RNA Helicase Family in Physic Nut (Jatropha curcas L.): Structural Characterization and Response to Salinity. Plants, 13(6), 905. https://doi.org/10.3390/plants13060905