
Transcriptome Analysis Reveals Novel Genes Potentially Involved in Tuberization in Potato

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
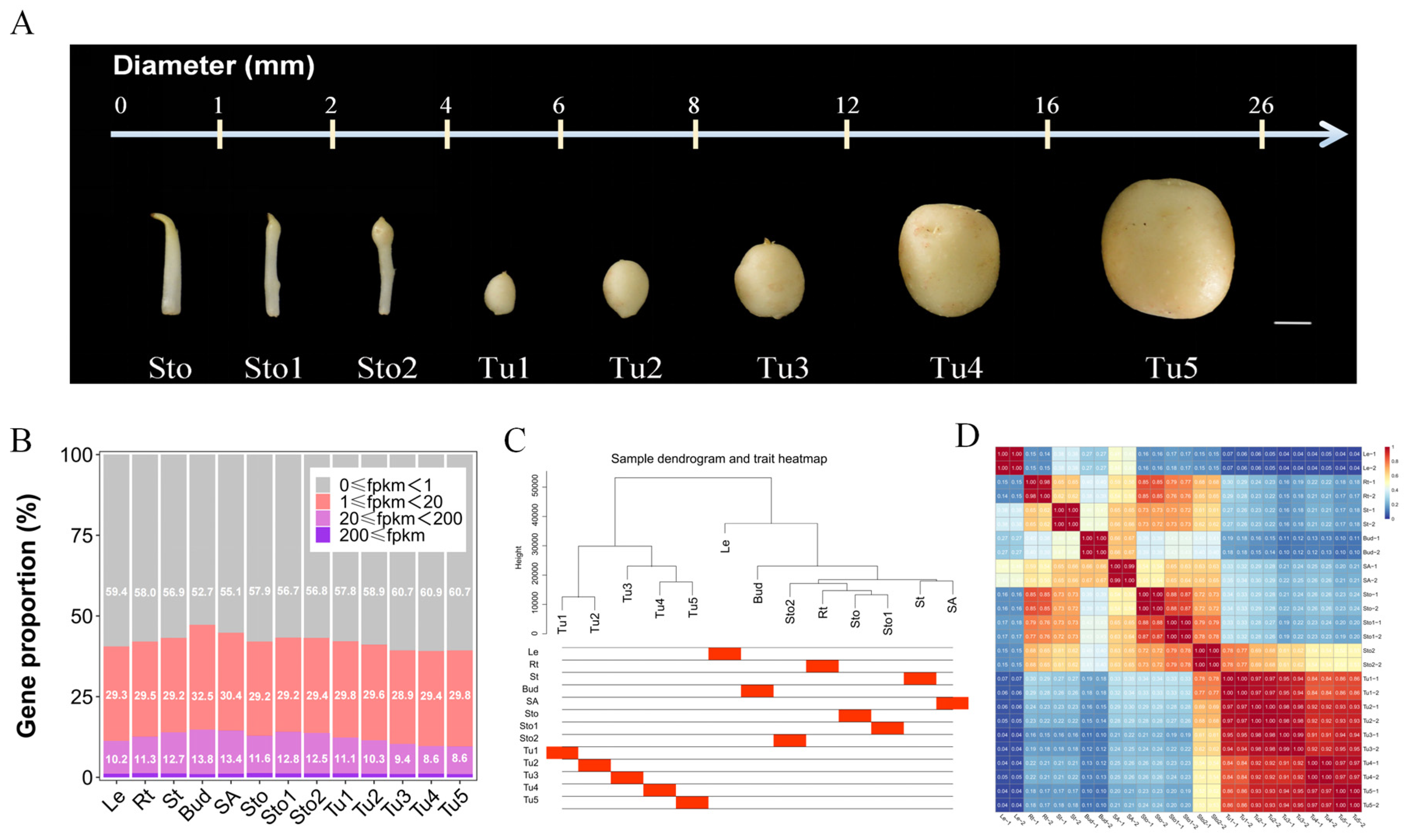
2.1. Global Analysis of Transcriptome
2.2. Co-Expression Networks Associated with Thirteen Tissues
2.3. DEGs and DETFs of Stolons and Tubers in Different Developmental Stages
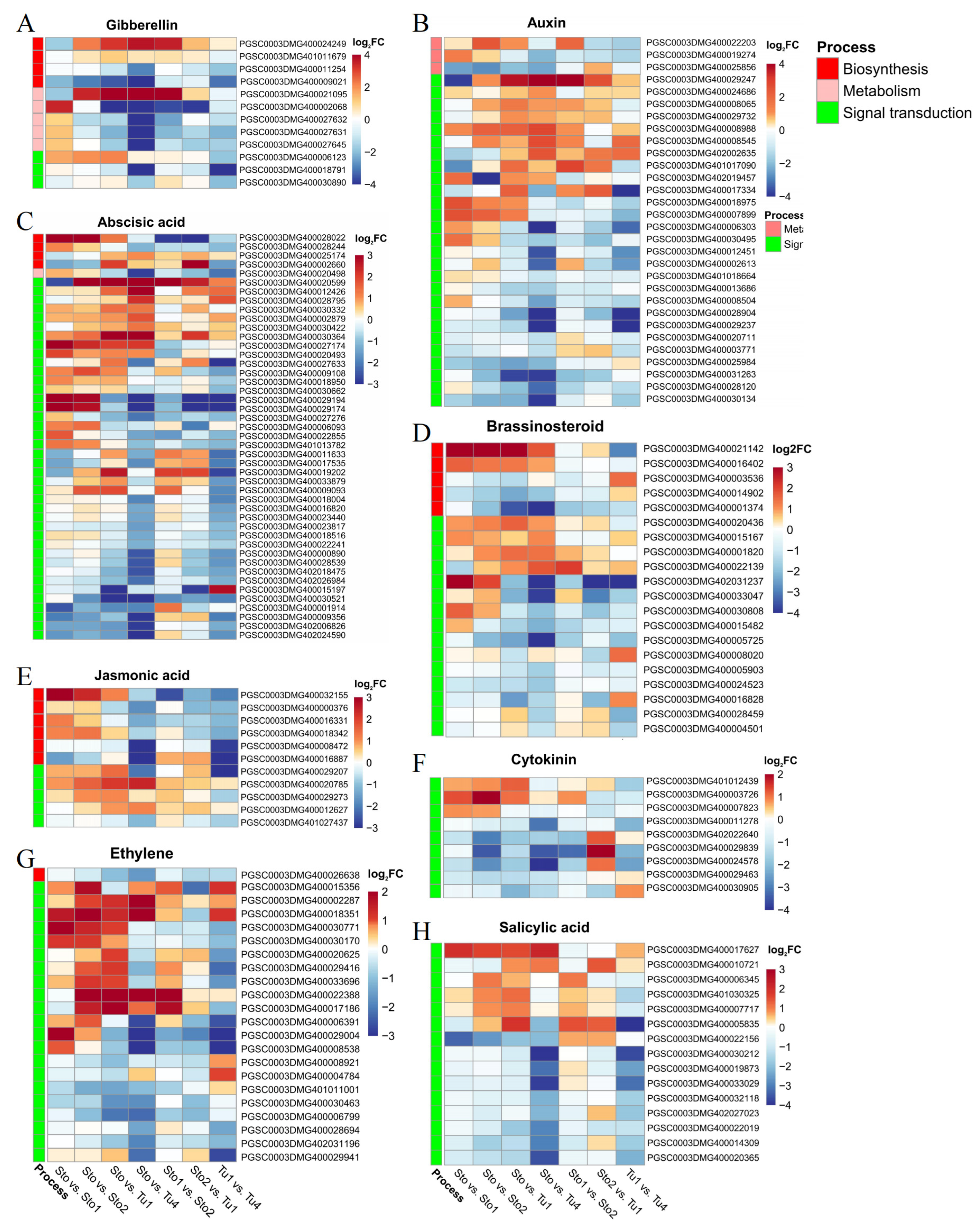
2.4. DEGs of Hormone Regulation Pathway
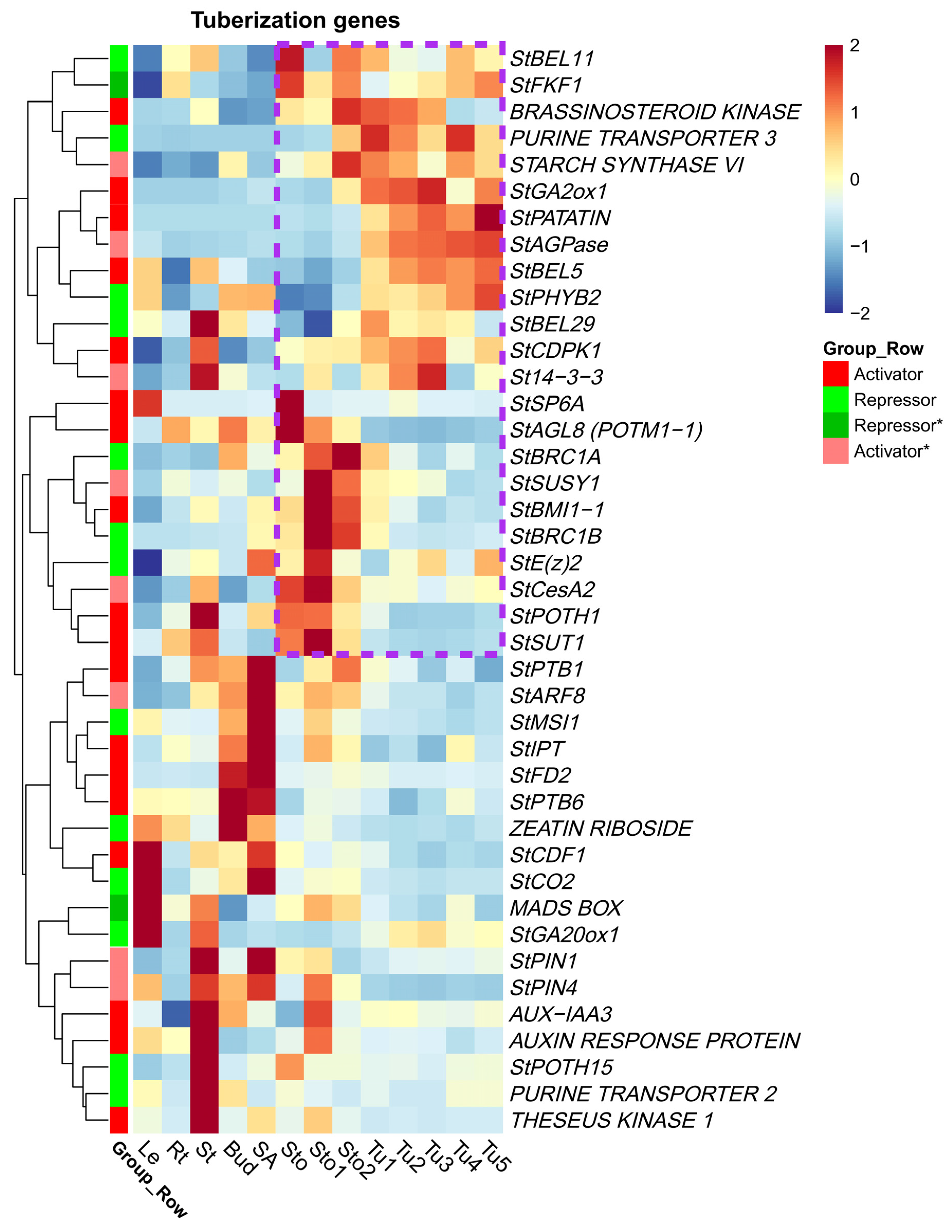
2.5. DEGs of Reported Tuberization Regulatory Network
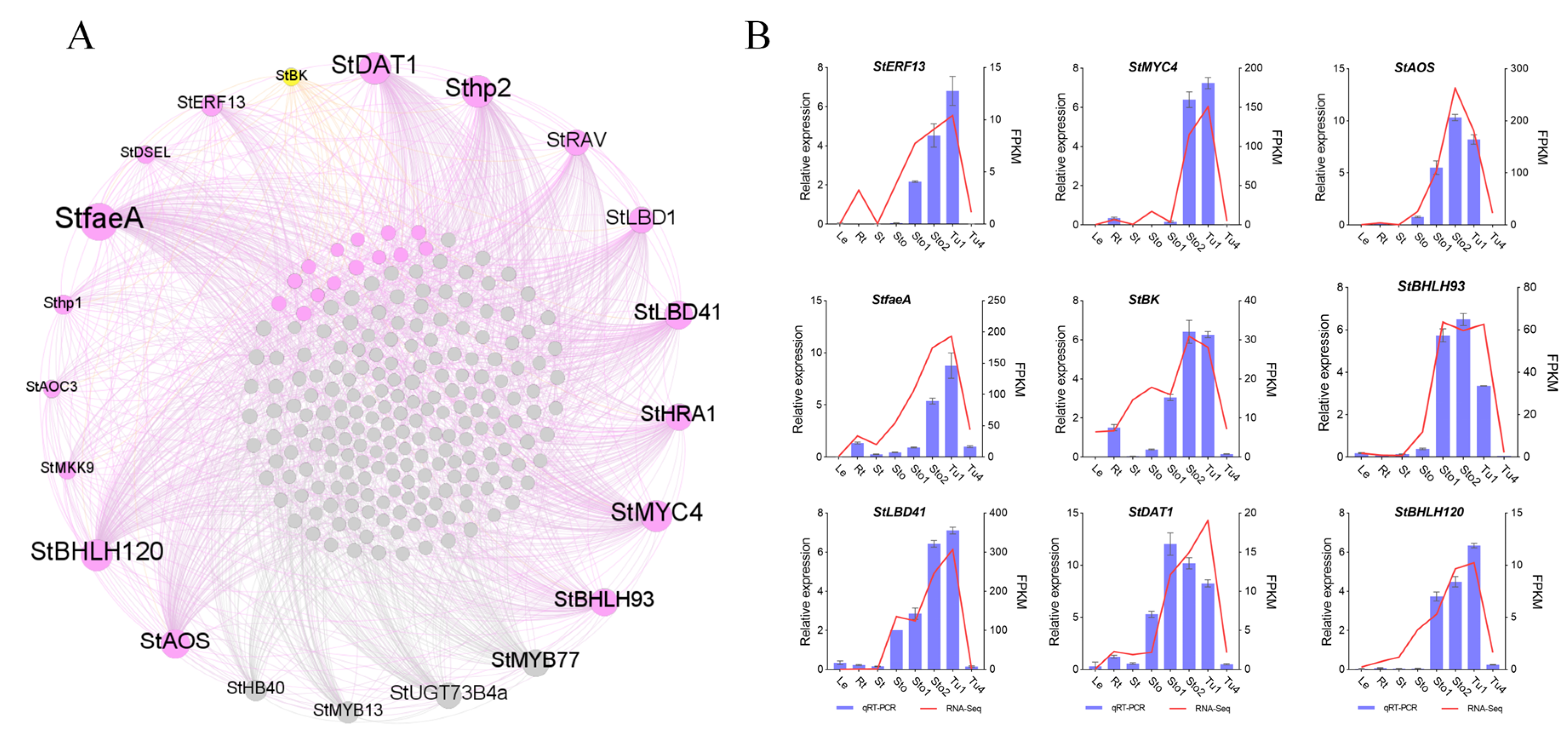
2.6. Co-Expression Networks’ Construction of Stolons and Tubers Associative Modules
3. Discussion
4. Materials and Methods
4.1. Materials and Growth Conditions
4.2. Total RNA Extraction and cDNA Library Construction
4.3. High-Throughput RNA-Seq and Data Analysis
4.4. Gene Functional Enrichment Analysis
4.5. Construction of Weighted Gene Co-Expression Networks
4.6. Real-Time Quantitative PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kondhare, K.R.; Natarajan, B.; Banerjee, A.K. Molecular signals that govern tuber development in potato. Int. J. Dev. Biol. 2020, 64, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Zierer, W.; Rüscher, D.; Sonnewald, U.; Sonnewald, S. Tuber and tuberous root development. Annu. Rev. Plant. Biol. 2021, 72, 551–580. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Jia, L.; Hu, B.; Alva, A.; Fan, M. Potato stolon and tuber growth influenced by nitrogen form. Plant Prod. Sci. 2014, 17, 138–143. [Google Scholar] [CrossRef]
- Ewing, E.E.; Struik, P.C. Tuber formation in potato: Induction, initiation, and growth. Horticult. Rev. 2010, 14, 89–198. [Google Scholar] [CrossRef]
- Kloosterman, B.; Navarro, C.; Bijsterbosch, G.; Lange, T.; Prat, S.; Visser, R.G.; Bachem, C.W. StGA2ox1 is induced prior to stolon swelling and controls GA levels during potato tuber development. Plant J. 2007, 52, 362–373. [Google Scholar] [CrossRef]
- Roumeliotis, E.; Kloosterman, B.; Oortwijn, M.; Kohlen, W.; Bouwmeester, H.J.; Visser, R.G.; Bachem, C.W. The effects of auxin and strigolactones on tuber initiation and stolon architecture in potato. J. Exp. Bot. 2012, 63, 4539–4547. [Google Scholar] [CrossRef]
- Tao, G.Q.; Stuart, D.; Yong, J.W.H.; Zhang, K.; Farquhar, G.D. Promotion of shoot development and tuberization in potato by expression of a chimaeric cytokinin synthesis gene at normal and elevated CO2 levels. Funct. Plant. Biol. 2010, 37, 43–54. [Google Scholar] [CrossRef]
- Gao, J.; Cao, X.; Shi, S.; Ma, Y.; Wang, K.; Liu, S.; Chen, D.; Chen, Q.; Ma, H. Genome-wide survey of Aux/IAA gene family members in potato (Solanum tuberosum): Identification, expression analysis, and evaluation of their roles in tuber development. Biochem. Biophys. Res. Commun. 2016, 471, 320–327. [Google Scholar] [CrossRef]
- Muñiz García, M.N.; Stritzler, M.; Capiati, D.A. Heterologous expression of Arabidopsis ABF4 gene in potato enhances tuberization through ABA-GA crosstalk regulation. Planta 2014, 239, 615–631. [Google Scholar] [CrossRef]
- Sarkar, D. The signal transduction pathways controlling in planta tuberization in potato: An emerging synthesis. Plant Cell Rep. 2008, 27, 1–8. [Google Scholar] [CrossRef]
- Kondhare, K.R.; Kumar, A.; Patil, N.S.; Malankar, N.N.; Saha, K.; Banerjee, A.K. Development of aerial and belowground tubers in potato is governed by photoperiod and epigenetic mechanism. Plant Physiol. 2021, 187, 1071–1086. [Google Scholar] [CrossRef] [PubMed]
- Dutt, S.; Manjul, A.S.; Raigond, P.; Singh, B.; Siddappa, S.; Bhardwaj, V.; Kawar, P.G.; Patil, V.U.; Kardile, H.B. Key players associated with tuberization in potato: Potential candidates for genetic engineering. Crit. Rev. Biotechnol. 2017, 37, 942–957. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, B.; De Koeyer, D.; Griffiths, R.; Flinn, B.; Steuernagel, B.; Scholz, U.; Sonnewald, S.; Sonnewald, U.; Bryan, G.J.; Prat, S.; et al. Genes driving potato tuber initiation and growth: Identification based on transcriptional changes using the POCI array. Funct. Integr. Genom. 2008, 8, 329–340. [Google Scholar] [CrossRef]
- Chen, H.; Banerjee, A.K.; Hannapel, D.J. The tandem complex of BEL and KNOX partners is required for transcriptional repression of ga20ox1. Plant J. 2004, 38, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Kondhare, K.R.; Patil, A.B.; Giri, A.P. Auxin: An emerging regulator of tuber and storage root development. Plant Sci. 2021, 306, 110854. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, M.; Torres-Pérez, R.; Wahl, V.; Cruz-Oró, E.; Rodríguez-Buey, M.L.; Zamarreño, A.M.; Martín-Jouve, B.; García-Mina, J.M.; Oliveros, J.C.; Prat, S.; et al. Spatial control of potato tuberization by the TCP transcription factor BRANCHED1b. Nat. Plants 2022, 8, 281–294. [Google Scholar] [CrossRef]
- Kühn, C.; Hajirezaei, M.R.; Fernie, A.R.; Roessner-Tunali, U.; Czechowski, T.; Hirner, B.; Frommer, W.B. The sucrose transporter StSUT1 localizes to sieve elements in potato tuber phloem and influences tuber physiology and development. Plant Physiol. 2003, 131, 102–113. [Google Scholar] [CrossRef]
- Zhao, P.; Liu, L.; Cao, J.; Wang, Z.; Zhao, Y.; Zhong, N. Transcriptome analysis of tryptophan-induced resistance against potato common scab. Int. J. Mol. Sci. 2022, 23, 8420. [Google Scholar] [CrossRef]
- Li, Q.; Qin, Y.; Hu, X.; Li, G.; Ding, H.; Xiong, X.; Wang, W. Transcriptome analysis uncovers the gene expression profile of salt-stressed potato (Solanum tuberosum L.). Sci. Rep. 2020, 10, 5411. [Google Scholar] [CrossRef]
- Wiesel, L.; Davis, J.L.; Milne, L.; Redondo Fernandez, V.; Herold, M.B.; Middlefell, W.J.; Morris, J.; Hedley, P.E.; Harrower, B.; Newton, A.C.; et al. A transcriptional reference map of defence hormone responses in potato. Sci. Rep. 2015, 5, 15229. [Google Scholar] [CrossRef]
- Vulavala, V.K.R.; Fogelman, E.; Faigenboim, A.; Shoseyov, O.; Ginzberg, I. The transcriptome of potato tuber phellogen reveals cellular functions of cork cambium and genes involved in periderm formation and maturation. Sci. Rep. 2019, 9, 10216. [Google Scholar] [CrossRef]
- Tiwari, J.K.; Buckseth, T.; Zinta, R.; Saraswati, A.; Singh, R.K.; Rawat, S.; Dua, V.K.; Chakrabarti, S.K. Transcriptome analysis of potato shoots, roots and stolons under nitrogen stress. Sci. Rep. 2020, 10, 1152. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Cai, Z.; Huang, J.; Wang, A.; Ntambiyukuri, A.; Chen, B.; Zheng, G.; Li, H.; Huang, Y.; Zhan, J.; et al. Transcriptomic analysis of tuberous root in two sweet potato varieties reveals the important genes and regulatory pathways in tuberous root development. BMC Genom. 2022, 23, 473. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Zhou, T.; Jing, S.; Dong, L.; Sun, X.; Fan, Y.; Shen, Y.; Liu, T.; Song, B. Leaves and stolons transcriptomic analysis provide insight into the role of phytochrome F in potato flowering and tuberization. Plant J. 2023, 113, 402–415. [Google Scholar] [CrossRef]
- Shan, J.; Song, W.; Zhou, J.; Wang, X.; Xie, C.; Gao, X.; Xie, T.; Liu, J. Transcriptome analysis reveals novel genes potentially involved in photoperiodic tuberization in potato. Genomics 2013, 102, 388–396. [Google Scholar] [CrossRef]
- Lu, Y.; Li, D.W.; Wu, Q.; Zhu, G.T. Transcriptome analysis of potato tubers at different developmental stages. J. Yunan Norm. Univ. Nat. Sci. Ed. 2021, 41, 25–32. (In Chinese) [Google Scholar]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Xu, X.; van Lammeren, A.A.; Vermeer, E.; Vreugdenhil, D. The role of gibberellin, abscisic acid, and sucrose in the regulation of potato tuber formation in vitro. Plant Physiol. 2005, 117, 575–584. [Google Scholar] [CrossRef]
- Sun, S.; Yi, C.; Ma, J.; Wang, S.; Peirats-Llobet, M.; Lewsey, M.G.; Whelan, J.; Shou, H. Analysis of spatio-temporal transcriptome profiles of soybean (Glycine max) tissues during early seed development. Int. J. Mol. Sci. 2020, 21, 7603. [Google Scholar] [CrossRef]
- Rashid, M.H.A.; Cheng, W.; Thomas, B. Temporal and spatial expression of Arabidopsis gene homologs control daylength adaptation and bulb formation in onion (Allium cepa L.). Sci. Rep. 2019, 9, 14629. [Google Scholar] [CrossRef]
- Hannapel, D.; Sharma, P.; Lin, T.; Banerjee, A.K. The multiple signals that control tuber formation. Plant Physiol. 2017, 174, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, E.; Visser, R.G.; Bachem, C.W. A crosstalk of auxin and GA during tuber development. Plant Signal Behav. 2012, 7, 1360–1363. [Google Scholar] [CrossRef] [PubMed]
- Abelenda, J.A.; Navarro, C.; Prat, S. Flowering and tuberization: A tale of two nightshades. Trends Plant Sci. 2014, 19, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Romano, J.M.; Dubos, C.; Prouse, M.B.; Wilkins, O.; Hong, H.; Poole, M.; Kang, K.Y.; Li, E.; Douglas, C.J.; Western, T.L.; et al. AtMYB61, an R2R3-MYB transcription factor, functions as a pleiotropic regulator via a small gene network. New Phytol. 2012, 195, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Reinders, A.; Panshyshyn, J.A.; Ward, J.M. Analysis of transport activity of Arabidopsis sugar alcohol permease homolog AtPLT5. J. Biol. Chem. 2005, 280, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Slabaugh, E.; Held, M.; Brandizzi, F. Control of root hair development in Arabidopsis thaliana by an endoplasmic reticulum anchored member of the R2R3-MYB transcription factor family. Plant J. 2005, 67, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Smit, M.E.; McGregor, S.R.; Sun, H.; Gough, C.; Bågman, A.M.; Soyars, C.L.; Kroon, J.T.; Gaudinier, A.; Williams, C.J.; Yang, X.; et al. A PXY-Mediated transcriptional network integrates signaling mechanisms to control vascular development in Arabidopsis. Plant Cell. 2020, 32, 319–335. [Google Scholar] [CrossRef]
- Riesmeier, J.W.; Hirner, B.; Frommer, W.B. Potato sucrose transporter expression in minor veins indicates a role in phloem loading. Plant Cell. 2020, 5, 1591–1598. [Google Scholar] [CrossRef]
- Graeff, M.; Straub, D.; Eguen, T.; Dolde, U.; Rodrigues, V.; Brandt, R.; Wenkel, S. MicroProtein-Mediated Recruitment of CONSTANS into a TOPLESS Trimeric Complex Represses Flowering in Arabidopsis. PLoS Genet. 2016, 12, e1005959. [Google Scholar] [CrossRef]
- Xu, M.; Li, X.; Xie, W.; Lin, C.; Wang, Q.; Tao, Z. ETHYLENE INSENSITIVE3/EIN3-LIKE1 modulate FLOWERING LOCUS C expression via histone demethylase interaction. Plant Physiol. 2023, 192, 2290–2300. [Google Scholar] [CrossRef]
- Chu, L.; Yang, C.; Zhuang, F.; Gao, Y.; Luo, M. The HDA9-HY5 module epigenetically regulates flowering time in Arabidopsis thaliana. J. Cell Physiol. 2023, 23, 2961–2968. [Google Scholar] [CrossRef]
- Wang, F.; Gao, Y.; Liu, Y.; Zhang, X.; Gu, X.; Ma, D.; Zhao, Z.; Yuan, Z.; Xue, H.; Liu, H. BES1-regulated BEE1 controls photoperiodic flowering downstream of blue light signaling pathway in Arabidopsis. New Phytol. 2019, 223, 1407–1419. [Google Scholar] [CrossRef]
- Shao, J.; Liu, X.; Wang, R.; Zhang, G.; Yu, F. The over-expression of an Arabidopsis B3 transcription factor, ABS2/NGAL1, leads to the loss of flower petals. PLoS ONE 2012, 7, e49861. [Google Scholar] [CrossRef]
- Jayawardhane, K.N.; Singer, S.D.; Ozga, J.A.; Rizvi, S.M.; Weselake, R.J.; Chen, G. Seed-specific down-regulation of Arabidopsis CELLULOSE SYNTHASE 1 or 9 reduces seed cellulose content and differentially affects carbon partitioning. Plant Cell Rep. 2020, 39, 953–969. [Google Scholar] [CrossRef]
- Stork, J.; Harris, D.; Griffiths, J.; Williams, B.; Beisson, F.; Li-Beisson, Y.; Mendu, V.; Haughn, G.; Debolt, S. CELLULOSE SYNTHASE9 serves a nonredundant role in secondary cell wall synthesis in Arabidopsis epidermal testa cells. Plant Physiol. 2010, 153, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Teeples, M.; Lin, L.; de Lucas, M.; Turco, G.; Toal, T.W.; Gaudinier, A.; Young, N.F.; Trabucco, G.M.; Veling, M.T.; Lamothe, R.; et al. An Arabidopsis gene regulatory network for secondary cell wall synthesis. Nature 2015, 517, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; Somssich, M.; Nakata, M.T.; Unda, F.; Atsuzawa, K.; Kaneko, Y.; Wang, T.; Bågman, A.M.; Gaudinier, A.; Yoshida, K.; et al. Complete substitution of a secondary cell wall with a primary cell wall in Arabidopsis. Nat. Plants 2018, 4, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zheng, C.; Zhao, Y.; Li, Q.; Liu, J.; Deng, R.; Lei, T.; Wang, S.; Wang, X. RNA interference knockdown of the brassinosteroid receptor BRI1 in potato (Solanum tuberosum L.) reveals novel functions for brassinosteroid signaling in controlling tuberization. Sci. Hortic. 2021, 290, 110516. [Google Scholar] [CrossRef]
- Sharma, N.; Xin, R.; Kim, D.H.; Sung, S.; Lange, T.; Huq, E. NO FLOWERING IN SHORT DAY (NFL) is a bHLH transcription factor that promotes flowering specifically under short-day conditions in Arabidopsis. Development 2016, 143, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Lackman, P.; González-Guzmán, M.; Tilleman, S.; Carqueijeiro, I.; Pérez, A.C.; Moses, T.; Seo, M.; Kanno, Y.; Häkkinen, S.T.; Van Montagu, M.C.; et al. Jasmonate signaling involves the abscisic acid receptor PYL4 to regulate metabolic reprogramming in Arabidopsis and tobacco. Proc. Natl. Acad. Sci. USA 2011, 108, 5891–5896. [Google Scholar] [CrossRef] [PubMed]
- Khokon, M.A.R.; Salam, M.A.; Jammes, F.; Ye, W.; Hossain, M.A.; Okuma, E.; Nakamura, Y.; Mori, I.C.; Kwak, J.M.; Murata, Y. MPK9 and MPK12 function in SA-induced stomatal closure in Arabidopsis thaliana. Biosci. Biotechnol. Biochem. 2017, 81, 1394–1400. [Google Scholar] [CrossRef]
- Liu, K.H.; Niu, Y.; Konishi, M.; Wu, Y.; Du, H.; Sun, C.H.; Li, L.; Boudsocq, M.; McCormack, M.; Maekawa, S.; et al. Discovery of nitrate-CPK-NLP signalling in central nutrient-growth networks. Nature 2017, 545, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Aliche, E.B.; Theeuwen, T.P.J.M.; Oortwijn, M.; Visser, R.G.F.; van der Linden, C.G. Carbon partitioning mechanisms in POTATO under drought stress. Plant Physiol. Biochem. 2020, 146, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Brust, H.; Lehmann, T.; D’Hulst, C.; Fettke, J. Analysis of the functional interaction of Arabidopsis starch synthase and branching enzyme isoforms reveals that the cooperative action of SSI and BEs results in glucans with polymodal chain length distribution similar to amylopectin. PLoS ONE 2017, 9, e102364. [Google Scholar] [CrossRef] [PubMed]
- Deniaud, A.; Panwar, P.; Frelet-Barrand, A.; Bernaudat, F.; Juillan-Binard, C.; Ebel, C.; Rolland, N.; Pebay-Peyroula, E. Oligomeric status and nucleotide binding properties of the plastid ATP/ADP transporter 1: Toward a molecular understanding of the transport mechanism. PLoS ONE 2012, 7, e32325. [Google Scholar] [CrossRef]
- Zeeman, S.C.; Thorneycroft, D.; Schupp, N.; Chapple, A.; Weck, M.; Dunstan, H.; Haldimann, P.; Bechtold, N.; Smith, A.M.; Smith, S.M. Plastidial alpha-glucan phosphorylase is not required for starch degradation in Arabidopsis leaves but has a role in the tolerance of abiotic stress. Plant Physiol. 2004, 135, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.; Guan, Z.; Wu, M.; Wang, X.; Fan, R.; Zhang, F.; Yan, J.; Liu, Y.; Zhang, D.; et al. The LSF1-MDH complex functions as a scaffold to recruit β-amylase to promote starch degradation. Plant Cell 2023, koad259. [Google Scholar] [CrossRef]
- Kevei, Z.; Baloban, M.; Da Ines, O.; Tiricz, H.; Kroll, A.; Regulski, K.; Mergaert, P.; Kondorosi, E. Conserved CDC20 cell cycle functions are carried out by two of the five isoforms in Arabidopsis thaliana. PLoS ONE 2011, 6, e20618. [Google Scholar] [CrossRef]
- Goldy, C.; Pedroza-Garcia, J.A.; Breakfield, N.; Cools, T.; Vena, R.; Benfey, P.N.; De Veylder, L.; Palatnik, J.; Rodriguez, R.E. The Arabidopsis GRAS-type SCL28 transcription factor controls the mitotic cell cycle and division plane orientation. Proc. Natl. Acad. Sci. USA 2021, 118, e2005256118. [Google Scholar] [CrossRef]
- Long, Y.; Smet, W.; Cruz-Ramírez, A.; Castelijns, B.; de Jonge, W.; Mähönen, A.P.; Bouchet, B.P.; Perez, G.S.; Akhmanova, A.; Scheres, B.; et al. Arabidopsis BIRD Zinc Finger Proteins Jointly Stabilize Tissue Boundaries by Confining the Cell Fate Regulator SHORT-ROOT and Contributing to Fate Specification. Plant Cell 2015, 27, 1185–1199. [Google Scholar] [CrossRef]
- Ullah, H.; Chen, J.G.; Temple, B.; Boyes, D.C.; Alonso, J.M.; Davis, K.R.; Ecker, J.R.; Jones, A.M. The beta-subunit of the Arabidopsis G protein negatively regulates auxin-induced cell division and affects multiple developmental processes. Plant Cell 2003, 15, 393–409. [Google Scholar] [CrossRef]
- Heim, M.A.; Jakoby, M.; Werber, M.; Martin, C.; Weisshaar, B.; Bailey, P.C. The basic helix-loop-helix transcription factor family in plants: A genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 2003, 20, 735–747. [Google Scholar] [CrossRef]
- Gaudinier, A.; Rodriguez-Medina, J.; Zhang, L.; Olson, A.; Liseron-Monfils, C.; Bågman, A.M.; Foret, J.; Abbitt, S.; Tang, M.; Li, B.; et al. Transcriptional regulation of nitrogen-associated metabolism and growth. Nature 2018, 563, 259–264. [Google Scholar] [CrossRef]
- Bell, E.M.; Lin, W.C.; Husbands, A.Y.; Yu, L.; Jaganatha, V.; Jablonska, B.; Mangeon, A.; Neff, M.M.; Girke, T.; Springer, P.S. Arabidopsis lateral organ boundaries negatively regulates brassinosteroid accumulation to limit growth in organ boundaries. Proc. Natl. Acad. Sci. USA 2012, 109, 21146–21151. [Google Scholar] [CrossRef]
- Kang, M.; Abdelmageed, H.; Lee, S.; Reichert, A.; Mysore, K.S.; Allen, R.D. AtMBP-1, an alternative translation product of LOS2, affects abscisic acid responses and is modulated by the E3 ubiquitin ligase AtSAP5. Plant J. 2013, 76, 481–493. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Bolser, D.; de Boer, J.; Sønderkær, M.; Amoros, W.; Carboni, M.F.; D’Ambrosio, J.M.; de la Cruz, G.; Di Genova, A.; Douches, D.S.; et al. Construction of reference chromosome-scale pseudomolecules for potato: Integrating the potato genome with genetic and physical maps. G3 2013, 3, 2031–2047. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the Third International Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, N.; Si, H.; Calderón-Urrea, A. Selection and validation of reference genes for RT-qPCR analysis in potato under abiotic stress. Plant Methods 2017, 13, 85. [Google Scholar] [CrossRef]
- Kõressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: Integrating masking of template sequence with primer design software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Jian, H.; Shang, L.; Wang, K.; Wen, S.; Li, Z.; Liu, R.; Jia, L.; Huang, Z.; Lyu, D. Transcriptome Analysis Reveals Novel Genes Potentially Involved in Tuberization in Potato. Plants 2024, 13, 795. https://doi.org/10.3390/plants13060795
Zhang M, Jian H, Shang L, Wang K, Wen S, Li Z, Liu R, Jia L, Huang Z, Lyu D. Transcriptome Analysis Reveals Novel Genes Potentially Involved in Tuberization in Potato. Plants. 2024; 13(6):795. https://doi.org/10.3390/plants13060795
Chicago/Turabian StyleZhang, Meihua, Hongju Jian, Lina Shang, Ke Wang, Shiqi Wen, Zihan Li, Rongrong Liu, Lijun Jia, Zhenlin Huang, and Dianqiu Lyu. 2024. "Transcriptome Analysis Reveals Novel Genes Potentially Involved in Tuberization in Potato" Plants 13, no. 6: 795. https://doi.org/10.3390/plants13060795
APA StyleZhang, M., Jian, H., Shang, L., Wang, K., Wen, S., Li, Z., Liu, R., Jia, L., Huang, Z., & Lyu, D. (2024). Transcriptome Analysis Reveals Novel Genes Potentially Involved in Tuberization in Potato. Plants, 13(6), 795. https://doi.org/10.3390/plants13060795