
The ldp1 Mutation Affects the Expression of Auxin-Related Genes and Enhances SAM Size in Rice

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Agronomic Traits Analysis of the Rice ldp1 Mutants
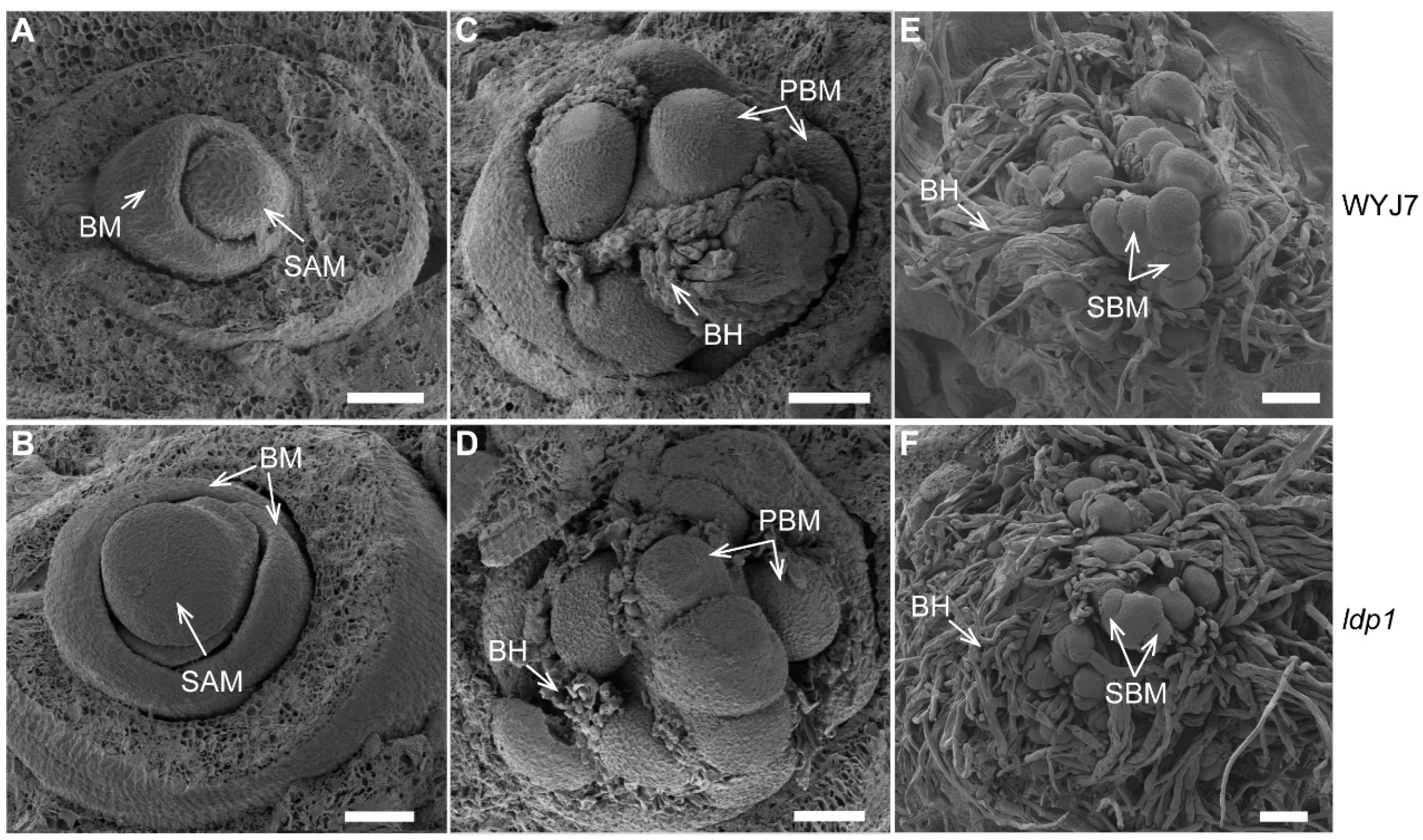
2.2. ldp1 Mutation Increases the Size of Meristem at the Apex of Inflorescence
2.3. RNA-Seq Analysis of the Three Initial Stages of Panicle Development from the SAM in WYJ7 and ldp1
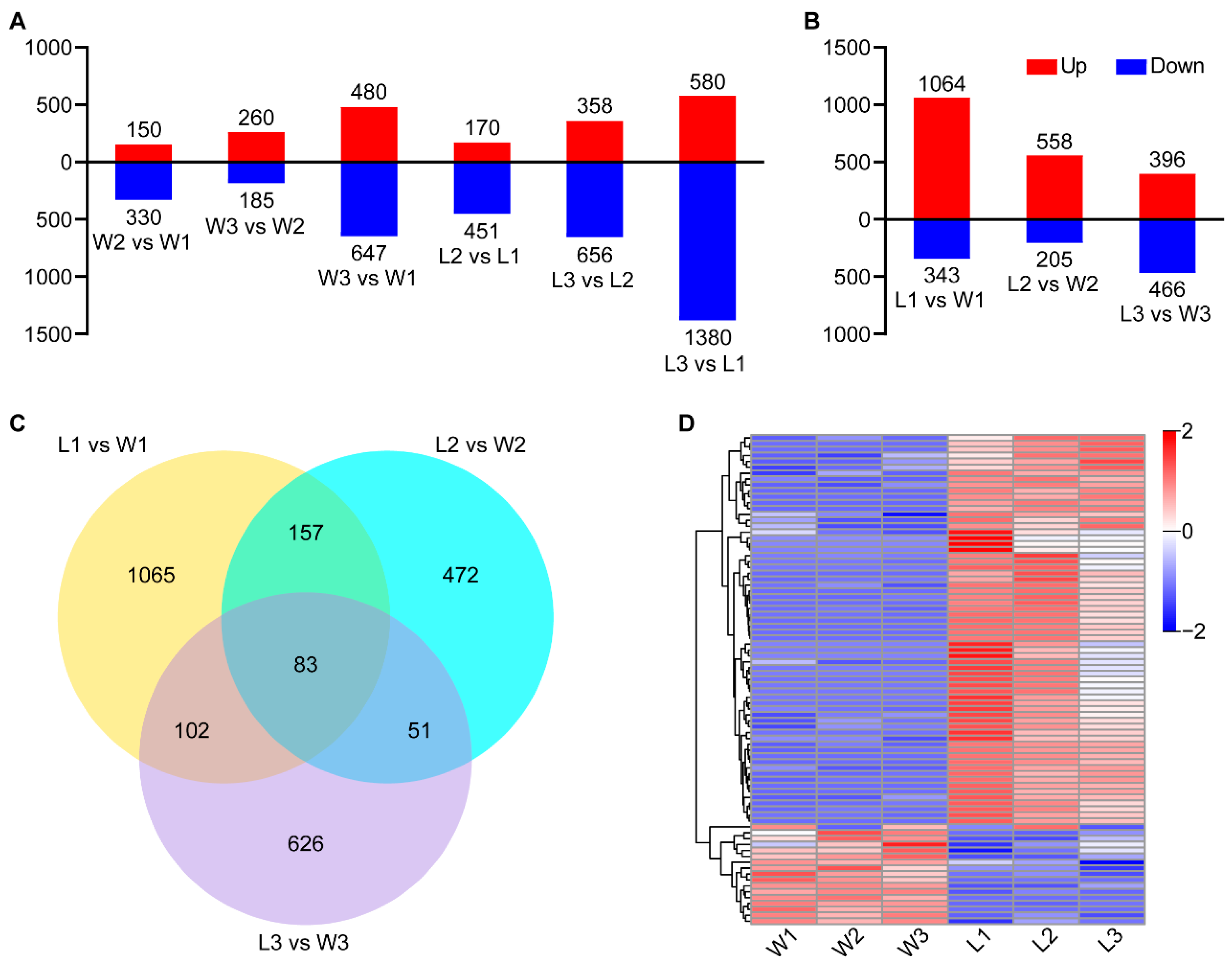
2.3.1. Transcriptome Sequencing and Analysis of Differentially Expressed Genes (DEGs)
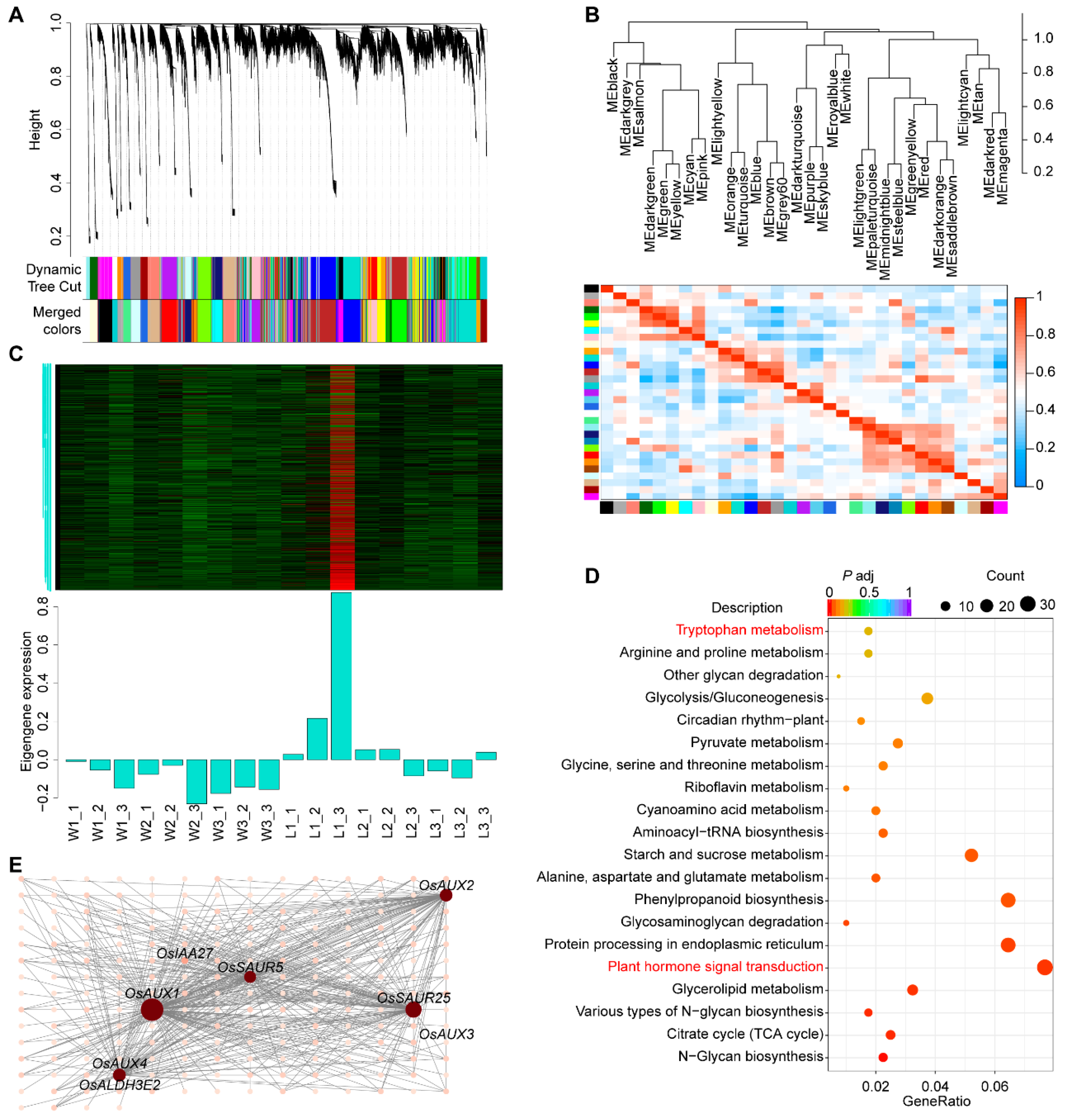
2.3.2. Analysis of WGCNA and Gene Coexpression Network
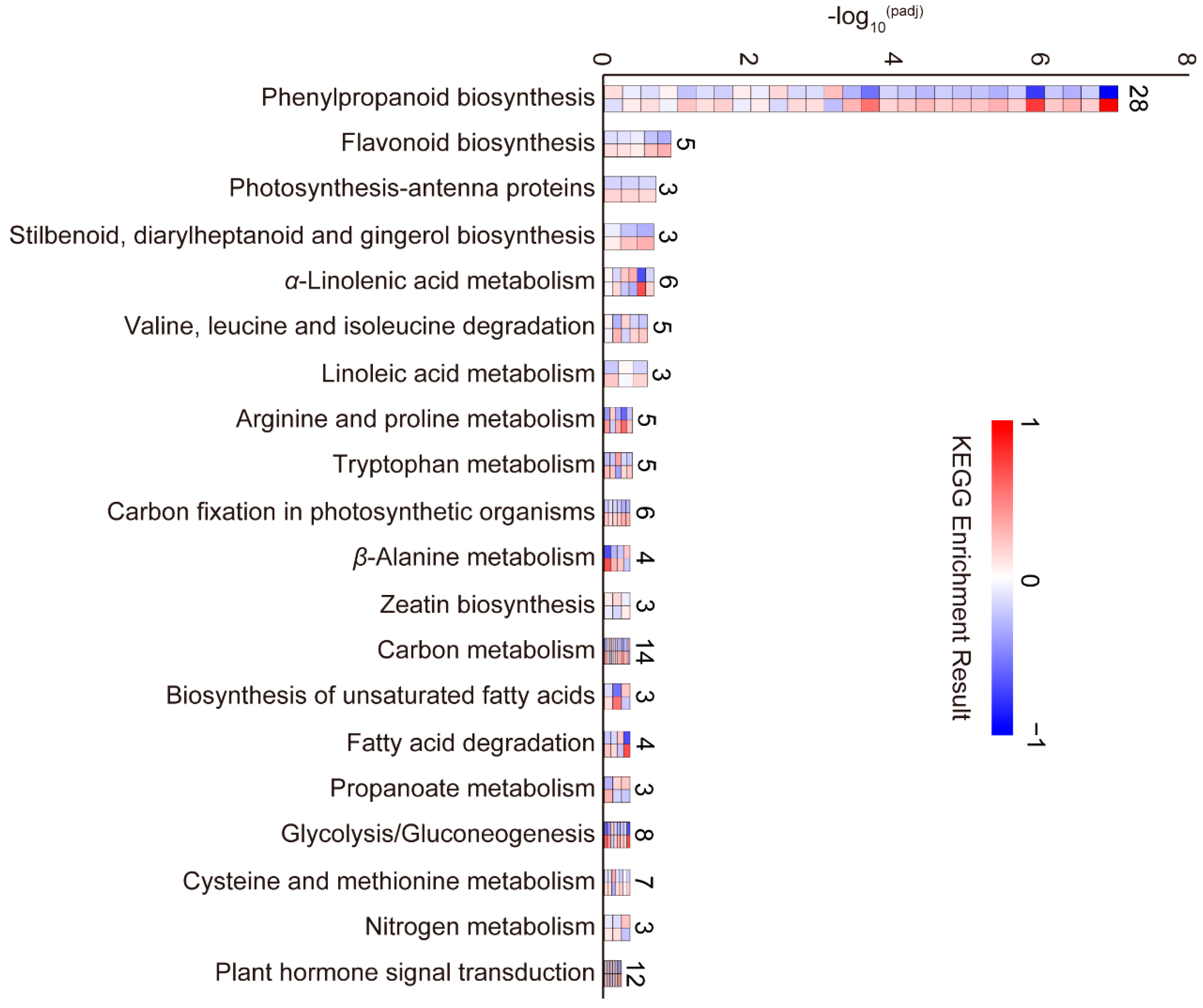
2.3.3. GO and KEGG Enrichment Analysis of DEGs during the BM Stage
2.4. Genetic Analysis of the Rice ldp1 Mutant Phenotypes and Gene Mapping of the LDP1 Gene
2.5. Analysis of the Candidate Genes
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Planting, Sampling, and Agronomic Trait Investigation of Rice Materials
5.2. Scanning Electron Microscopy
5.3. RNA-Seq and WGCNA Analyses
5.4. Quantitative Real-Time PCR (qRT-PCR) Analysis
5.5. Construction of F2 Segregating Population and Mapping of the LDP1 Gene
5.6. Candidate Gene Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sakamoto, T.; Matsuoka, M. Identifying and exploiting grain yield genes in rice. Curr. Opin. Plant Biol. 2008, 11, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.Z.; Zhang, Q.F. Genetic and molecular bases of rice yield. Annu. Rev. Plant Biol. 2010, 61, 421–442. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.J.; Hua, K.; Xu, R.; Zeng, D.L.; Wang, R.C.; Dong, G.J.; Zhang, G.Z.; Lu, X.L.; Fang, N.; Wang, D.K.; et al. The LARGE2-APO1/APO2 regulatory module controls panicle size and grain number in rice. Plant Cell 2021, 33, 1212–1228. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Li, J.Y. Molecular basis of plant architecture. Annu. Rev. Plant Biol. 2008, 59, 253–279. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.; Kumar, A.; Li, X.Y. Genetic and molecular pathways controlling rice inflorescence architecture. Front. Plant Sci. 2022, 13, 1010138. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Sunohara, H.; Nagato, Y. Developmental course of inflorescence and spikelet in rice. Breed. Sci. 2004, 54, 147–156. [Google Scholar] [CrossRef]
- Wang, Y.L.; Bi, X.J.; Zhong, J.S. Revisiting the origin and identity specification of the spikelet: A structural innovation in grasses (Poaceae). Plant Physiol. 2022, 190, 60–71. [Google Scholar] [CrossRef]
- Komatsu, M.; Maekawa, M.; Shimamoto, K.; Kyozuka, J. The LAX1 and FRIZZY PANICLE 2 genes determine the inflorescence architecture of rice by controlling rachis-branch and spikelet development. Dev. Biol. 2001, 231, 364–373. [Google Scholar] [CrossRef]
- Komatsu, K.; Maekawa, M.; Ujiie, S.; Satake, Y.; Furutani, I.; Okamoto, H.; Shimamoto, K.; Kyozuka, J. LAX and SPA: Major regulators of shoot branching in rice. Proc. Natl. Acad. Sci. USA 2003, 100, 11765–11770. [Google Scholar] [CrossRef]
- Tabuchi, H.; Zhang, Y.; Hattori, S.; Omae, M.; Shimizu-Sato, S.; Oikawa, T.; Qian, Q.; Nishimura, M.; Kitano, H.; Xie, H.; et al. LAX PANICLE2 of rice encodes a novel nuclear protein and regulates the formation of axillary meristems. Plant Cell 2011, 23, 3276–3287. [Google Scholar] [CrossRef]
- Tsuda, K.; Ito, Y.; Sato, Y.; Kurata, N. Positive autoregulation of a KNOX gene is essential for shoot apical meristem maintenance in rice. Plant Cell 2011, 23, 4368–4381. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Nagasawa, N.; Nagato, Y. ABERRANT PANICLE ORGANIZATION 1 temporally regulates meristem identity in rice. Dev. Biol. 2005, 282, 349–360. [Google Scholar] [CrossRef]
- Ikeda-Kawakatsu, K.; Yasuno, N.; Oikawa, T.; Iida, S.; Nagato, Y.; Maekawa, M.; Kyozuka, J. Expression level of ABERRANT PANICLE ORGANIZATION1 determines rice inflorescence form through control of cell proliferation in the meristem. Plant Physiol. 2009, 50, 736–747. [Google Scholar] [CrossRef]
- Rao, N.N.; Prasad, K.; Kumar, P.R.; Vijayraghavan, U. Distinct regulatory role for RFL, the rice LFY homolog, in determining flowering time and plant architecture. Proc. Natl. Acad. Sci. USA 2008, 105, 3646–3651. [Google Scholar] [CrossRef] [PubMed]
- Ikeda-Kawakatsu, K.; Maekawa, M.; Izawa, T.; Itoh, J.; Nagato, Y. ABERRANT PANICLE ORGANIZATION 2/RFL, the rice ortholog of Arabidopsis LEAFY, suppresses the transition from inflorescence meristem to floral meristem through interaction with APO1. Plant J. 2012, 69, 168–180. [Google Scholar] [CrossRef]
- Miao, Y.L.; Xun, Q.; Taji, T.; Tanaka, K.; Yasuno, N.; Ding, C.; Kyozuka, J. ABERRANT PANICLE ORGANIZATION2 controls multiple steps in panicle formation through common direct-target genes. Plant Physiol. 2022, 189, 2210–2226. [Google Scholar] [CrossRef]
- Yoshida, A.; Ohmori, Y.; Kitano, H.; Taguchi-Shiobara, F.; Hirano, H.Y. ABERRANT SPIKELET AND PANICLE1, encoding a TOPLESS-related transcriptional corepressor, is involved in the regulation of meristem fate in rice. Plant J. 2012, 70, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.; Fang, J.J.; Savelieva, E.M.; Lomin, S.N.; Shang, J.Y.; Sun, Y.L.; Zhao, J.F.; Kumar, A.; Yuan, S.J.; Yao, X.F.; et al. The cytokinin receptor OHK4/OsHK4 regulates inflorescence architecture in rice via an IDEAL PLANT ARCHITECTURE1/WEALTHY FARMER’S PANICLE-mediated positive feedback circuit. Plant Cell 2023, 36, 40–64. [Google Scholar] [CrossRef]
- Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef]
- Huang, X.Z.; Qian, Q.; Liu, Z.B.; Sun, H.Y.; He, S.Y.; Luo, D.; Xia, G.M.; Chu, C.C.; Li, J.Y.; Fu, X.D. Natural variation at the DEP1 locus enhances grain yield in rice. Nat. Genet. 2009, 41, 494–497. [Google Scholar] [CrossRef]
- Komatsu, M.; Chujo, A.; Nagato, Y.; Shimamoto, K.; Kyozuka, J. FRIZZY PANICLE is required to prevent the formation of axillary meristems and to establish floral meristem identity in rice spikelets. Development 2003, 130, 3841–3850. [Google Scholar] [CrossRef]
- Bai, X.F.; Huang, Y.; Mao, D.H.; Wen, M.; Zhang, L.; Xing, Y.Z. Regulatory role of FZP in the determination of panicle branching and spikelet formation in rice. Sci. Rep. 2016, 6, 19022. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.F.; Zhao, Y.D. A role for Auxin in flower development. J. Integr. Plant Biol. 2007, 49, 99–104. [Google Scholar] [CrossRef]
- Zhao, Y.D. Auxin biosynthesis and its role in plant development. Annu. Rev. Plant Biol. 2010, 61, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Deveshwar, P.; Prusty, A.; Sharma, S.; Tyagi, A.K. Phytohormone-Mediated Molecular Mechanisms Involving Multiple Genes and QTL Govern Grain Number in Rice. Front. Genet. 2020, 11, 586462. [Google Scholar] [CrossRef]
- Gälweiler, L.; Guan, C.; Müller, A.; Wisman, E.; Mendgen, K.; Yephremov, A.; Palme, K. Regulation of polar auxin transport by AtPIN1 in Arabidopsis vascular tissue. Science 1998, 282, 2226–2230. [Google Scholar] [CrossRef]
- Friml, J.; Vieten, A.; Sauer, M.; Weijers, D.; Schwarz, H.; Hamann, T.; Offringa, R.; Jürgens, G. Efflux-dependent auxin gradients establish the apical-basal axis of Arabidopsis. Nature 2003, 426, 147–153. [Google Scholar] [CrossRef]
- Lu, G.W.; Coneva, V.; Casaretto, J.A.; Ying, S.; Mahmood, K.; Liu, F.; Nambara, E.; Bi, Y.M.; Rothstein, S.J. OsPIN5b modulates rice (Oryza sativa) plant architecture and yield by changing auxin homeostasis, transport and distribution. Plant J. 2015, 83, 913–925. [Google Scholar] [CrossRef]
- Liu, J.J.; Shi, X.A.; Chang, Z.Y.; Ding, Y.G.; Ding, C.Q. Auxin Efflux Transporters OsPIN1c and OsPIN1d Function Redundantly in Regulating Rice (Oryza sativa L.) Panicle Development. Plant Cell Physiol. 2022, 63, 305–316. [Google Scholar] [CrossRef]
- Wu, H.M.; Xie, D.J.; Tang, Z.S.; Shi, D.Q.; Yang, W.C. PINOID regulates floral organ development by modulating auxin transport and interacts with MADS16 in rice. Plant Biotechnol. J. 2020, 18, 1778–1795. [Google Scholar] [CrossRef]
- Mravec, J.; Skůpa, P.; Bailly, A.; Hoyerová, K.; Krecek, P.; Bielach, A.; Petrásek, J.; Zhang, J.; Gaykova, V.; Stierhof, Y.D.; et al. Subcellular homeostasis of phytohormone auxin is mediated by the ER-localized PIN5 transporter. Nature 2009, 459, 1136–1140. [Google Scholar] [CrossRef]
- Zong, J.; Wang, L.; Zhu, L.; Bian, L.L.; Zhang, B.; Chen, X.F.; Huang, G.Q.; Zhang, X.L.; Fan, J.Y.; Cao, L.M.; et al. A rice single cell transcriptomic atlas defines the developmental trajectories of rice floret and inflorescence meristems. New Phytol. 2022, 234, 494–512. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Khush, G.S. Breaking the yield frontier of rice. GeoJournal 1995, 35, 329–332. [Google Scholar] [CrossRef]
- Jiao, Y.Q.; Wang, Y.H.; Xue, D.W.; Wang, J.; Yan, M.X.; Liu, G.F.; Dong, G.J.; Zeng, D.L.; Lu, Z.F.; Zhu, X.D.; et al. Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 2010, 42, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Viana, V.E.; Pegoraro, C.; Busanello, C.; Costa de Oliveira, A. Mutagenesis in Rice: The Basis for Breeding a New Super Plant. Front Plant Sci. 2019, 10, 1326. [Google Scholar] [CrossRef]
- Schoof, H.; Lenhard, M.; Haecker, A.; Mayer, K.F.; Jürgens, G.; Laux, T. The stem cell population of Arabidopsis shoot meristems in maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell 2000, 100, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Aichinger, E.; Kornet, N.; Friedrich, T.; Laux, T. Plant stem cell niches. Annu. Rev. Plant Biol. 2012, 63, 615–636. [Google Scholar] [CrossRef]
- Somssich, M.; Je, B.I.; Simon, R.; Jackson, D. CLAVATA-WUSCHEL signaling in the shoot meristem. Development 2016, 143, 3238–3248. [Google Scholar] [CrossRef]
- Chu, H.W.; Qian, Q.; Liang, W.Q.; Yin, C.S.; Tan, H.X.; Yao, X.; Yuan, Z.; Yang, J.; Huang, H.; Luo, D.; et al. The floral organ number4 gene encoding a putative ortholog of Arabidopsis CLAVATA3 regulates apical meristem size in rice. Plant Physiol. 2006, 142, 1039–1052. [Google Scholar] [CrossRef]
- Moon, S.; Jung, K.H.; Lee, D.E.; Lee, D.Y.; Lee, J.; An, K.; Kang, H.G.; An, G. The rice FON1 gene controls vegetative and reproductive development by regulating shoot apical meristem size. Mol. Cells 2006, 21, 147–152. [Google Scholar] [CrossRef]
- Suzaki, T.; Toriba, T.; Fujimoto, M.; Tsutsumi, N.; Kitano, H.; Hirano, H.Y. Conservation and diversification of meristem maintenance mechanism in Oryza sativa: Function of the FLORAL ORGAN NUMBER2 gene. Plant Cell Physiol. 2006, 47, 1591–1602. [Google Scholar] [CrossRef]
- Ohmori, Y.; Tanaka, W.; Kojima, M.; Sakakibara, H.; Hirano, H.Y. WUSCHEL-RELATED HOMEOBOX4 is involved in meristem maintenance and is negatively regulated by the CLE gene FCP1 in rice. Plant Cell 2013, 25, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Kurakawa, T.; Ueda, N.; Maekawa, M.; Kobayashi, K.; Kojima, M.; Nagato, Y.; Sakakibara, H.; Kyozuka, J. Direct control of shoot meristem activity by a cytokinin-activating enzyme. Nature 2007, 445, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Hong, S.K.; Tagiri, A.; Kitano, H.; Yamamoto, N.; Nagato, Y.; Matsuoka, M. A rice homeobox gene, OSH1, is expressed before organ differentiation in a specific region during early embryogenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 8117–8122. [Google Scholar] [CrossRef] [PubMed]
- Sentoku, N.; Sato, Y.; Kurata, N.; Ito, Y.; Kitano, H.; Matsuoka, M. Regional expression of the rice KN1-type homeobox gene family during embryo, shoot, and flower development. Plant Cell 1999, 11, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.L.; Sun, C.D.; Shen, C.J.; Wang, S.K.; Liu, F.; Liu, Y.; Chen, Y.L.; Li, C.Y.; Qian, Q.; Aryal, B.; et al. The auxin transporter, OsAUX1, is involved in primary root and root hair elongation and in Cd stress responses in rice (Oryza sativa L.). Plant J. 2015, 83, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Qiao, J.; Yu, C.; Chen, H.; Sun, C.; Huang, L.; Li, C.; Geisler, M.; Qian, Q.; Jiang, A.; et al. The auxin influx carrier, OsAUX3, regulates rice root development and responses to aluminium stress. Plant Cell Environ. 2019, 42, 1125–1138. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.G.; Wu, Y.R.; Gao, Z.Y.; Chen, H.; Jia, L.X.; Li, D.M.; Li, X.G.; Qian, Q.; Qi, Y.H. Primary root and root hair development regulation by OsAUX4 and its participation in the phosphate starvation response. J. Integr. Plant Biol. 2021, 63, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.Y.; Jiang, H.Z.; Lin, Y.Q.; Shang, L.G.; Wang, M.; Li, D.M.; Fu, X.D.; Geisler, M.; Qi, Y.H.; Gao, Z.Y.; et al. A novel miR167a-OsARF6-OsAUX3 module regulates grain length and weight in rice. Mol. Plant 2021, 14, 1683–1698. [Google Scholar] [CrossRef]
- Ren, H.; Gray, W.M. SAUR proteins as effectors of hormonal and environmental signals in plant growth. Mol. Plant 2015, 8, 1153–1164. [Google Scholar] [CrossRef]
- Song, Y.L.; Wang, L.; Xiong, L.Z. Comprehensive expression profiling analysis of OsIAA gene family in developmental processes and in response to phytohormone and stress treatments. Planta 2009, 229, 577–591. [Google Scholar] [CrossRef]
- Weijers, D.; Wagner, D. Transcriptional Responses to the Auxin Hormone. Annu. Rev. Plant Biol. 2016, 67, 539–574. [Google Scholar] [CrossRef]
- Du, M.; Spalding, E.P.; Gray, W.M. Rapid Auxin-Mediated Cell Expansion. Annu. Rev. Plant Biol. 2020, 71, 379–402. [Google Scholar] [CrossRef]
- Sun, Z.L.; Mei, T.R.; Feng, T.T.; Ai, H.; Ye, Y.F.; Duan, S.M.; Liu, B.M.; Huang, X.Z. Deletion of the OsLA1 Gene Leads to Multi-Tillering and Lazy Phenotypes in Rice. Agriculture 2023, 13, 2166. [Google Scholar] [CrossRef]
- Zhang, Y.; Park, C.; Bennett, C.; Thornton, M.; Kim, D. Rapid and accurate alignment of nucleotide conversion sequencing reads with HISAT-3N. Genome Res. 2021, 31, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.F.; Jin, Y.H.; Huang, W.; Sun, Q.; Liu, F.; Huang, X.Z. Full-length transcriptome sequences of ephemeral plant Arabidopsis pumila provides insight into gene expression dynamics during continuous salt stress. BMC Genom. 2018, 19, 717. [Google Scholar] [CrossRef]
- Ding, R.Y.; Qu, Y.Y.; Wu, C.H.; Vijay-Shanker, K. Automatic gene annotation using GO terms from cellular component domain. BMC Medical Inform. Decis. Mak. 2018, 18, 119. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.; Johnstone, C.; Thompson, C. A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res. 1991, 19, 1349. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Z.; Mei, T.; Tan, X.; Feng, T.; Li, R.; Duan, S.; Zhao, H.; Ye, Y.; Liu, B.; Zhou, A.; et al. The ldp1 Mutation Affects the Expression of Auxin-Related Genes and Enhances SAM Size in Rice. Plants 2024, 13, 759. https://doi.org/10.3390/plants13060759
Sun Z, Mei T, Tan X, Feng T, Li R, Duan S, Zhao H, Ye Y, Liu B, Zhou A, et al. The ldp1 Mutation Affects the Expression of Auxin-Related Genes and Enhances SAM Size in Rice. Plants. 2024; 13(6):759. https://doi.org/10.3390/plants13060759
Chicago/Turabian StyleSun, Zhanglun, Tianrun Mei, Xuan Tan, Tingting Feng, Ruining Li, Sumei Duan, Heming Zhao, Yafeng Ye, Binmei Liu, Aifeng Zhou, and et al. 2024. "The ldp1 Mutation Affects the Expression of Auxin-Related Genes and Enhances SAM Size in Rice" Plants 13, no. 6: 759. https://doi.org/10.3390/plants13060759
APA StyleSun, Z., Mei, T., Tan, X., Feng, T., Li, R., Duan, S., Zhao, H., Ye, Y., Liu, B., Zhou, A., Ai, H., & Huang, X. (2024). The ldp1 Mutation Affects the Expression of Auxin-Related Genes and Enhances SAM Size in Rice. Plants, 13(6), 759. https://doi.org/10.3390/plants13060759