
Transcriptome Profiling Reveals the Gene Network Responding to Low Nitrogen Stress in Wheat


 ,
,
Abstract
1. Introduction
2. Results
2.1. Wheat Seedling Morphology under Low Nitrogen Stress
2.2. Transcriptomics Quality and Mapping Statistics
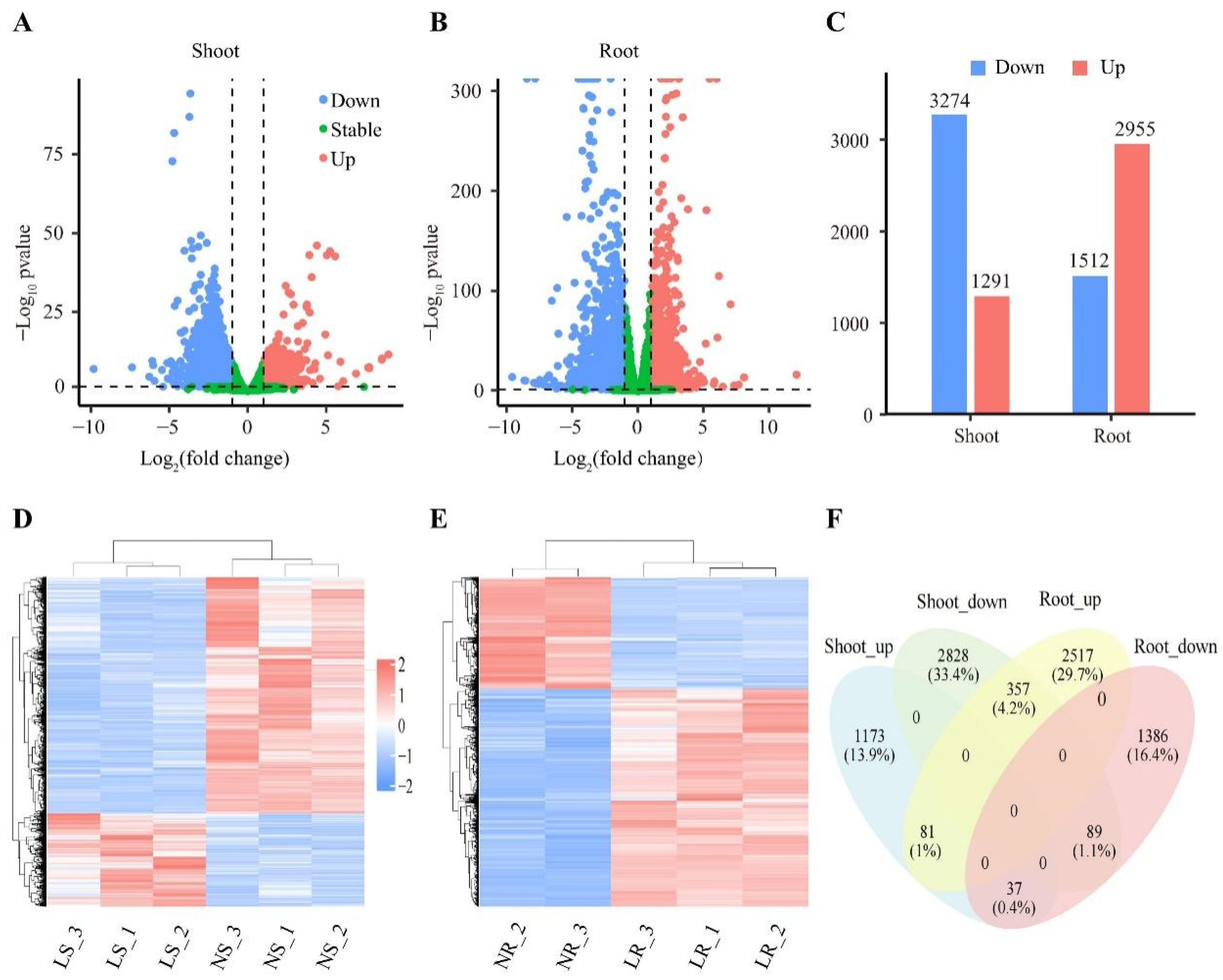
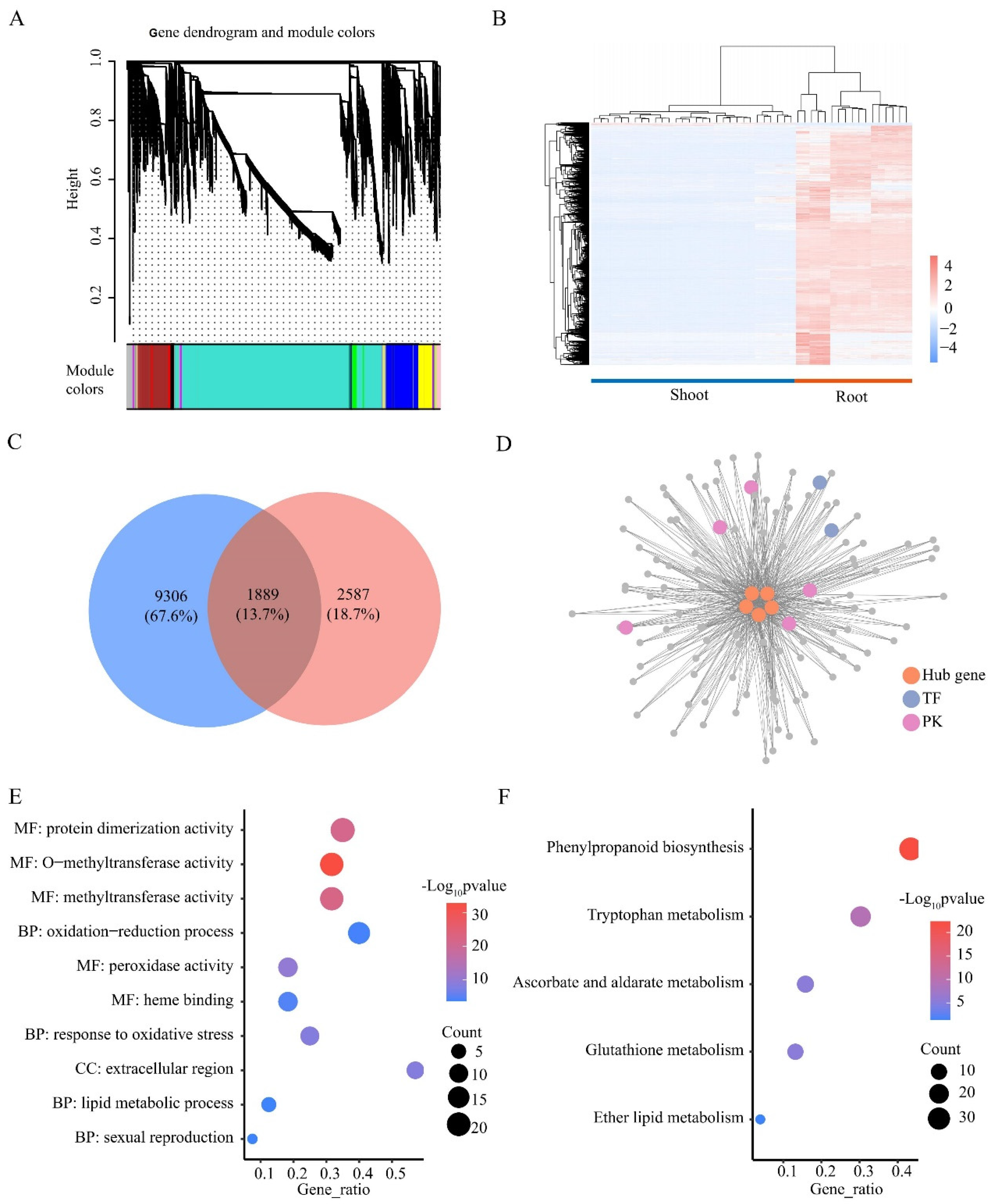
2.3. Identification and Analysis of Differentially Expressed Genes
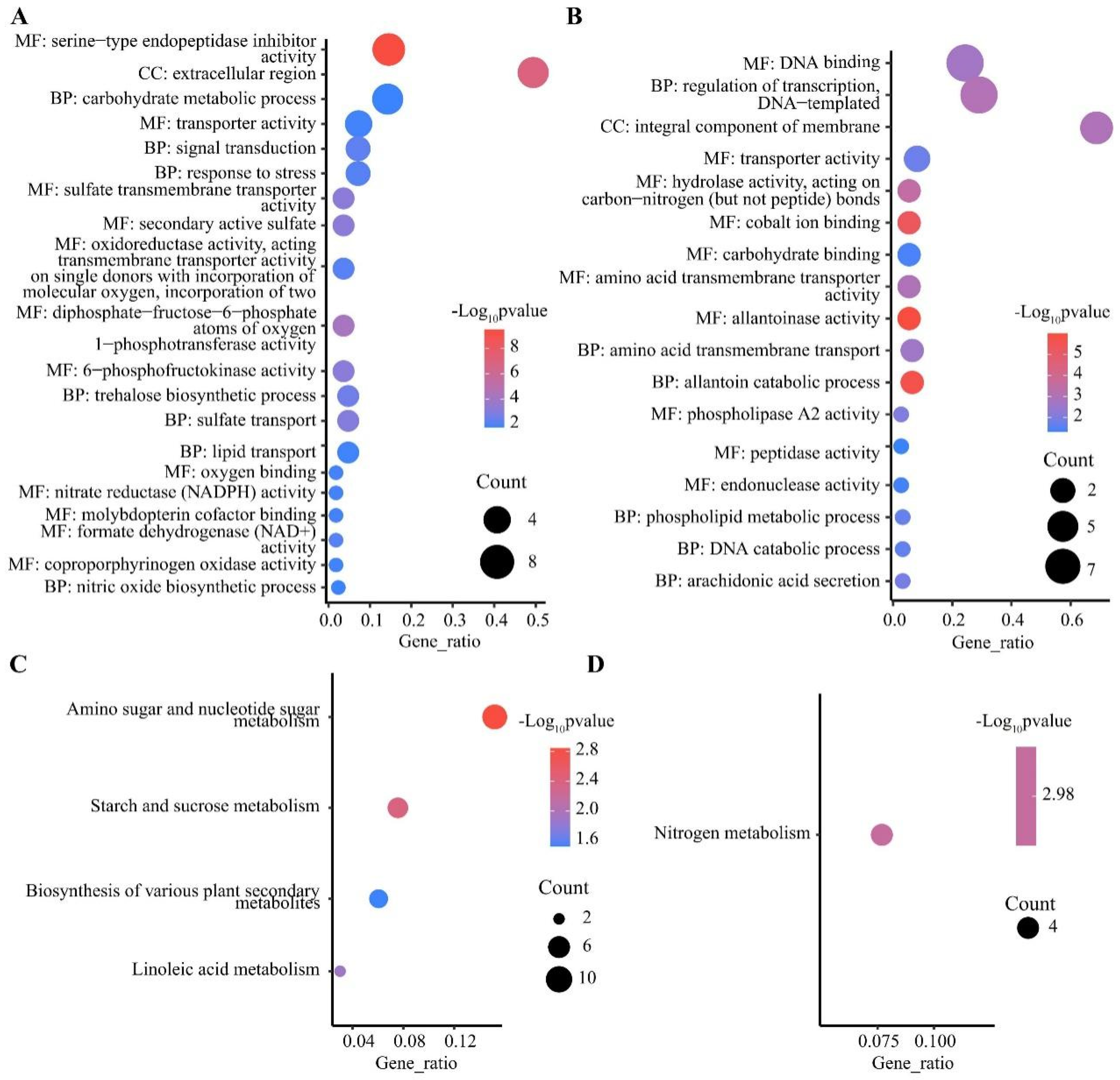
2.4. GO and KEGG Enrichment of Differentially Expressed Genes
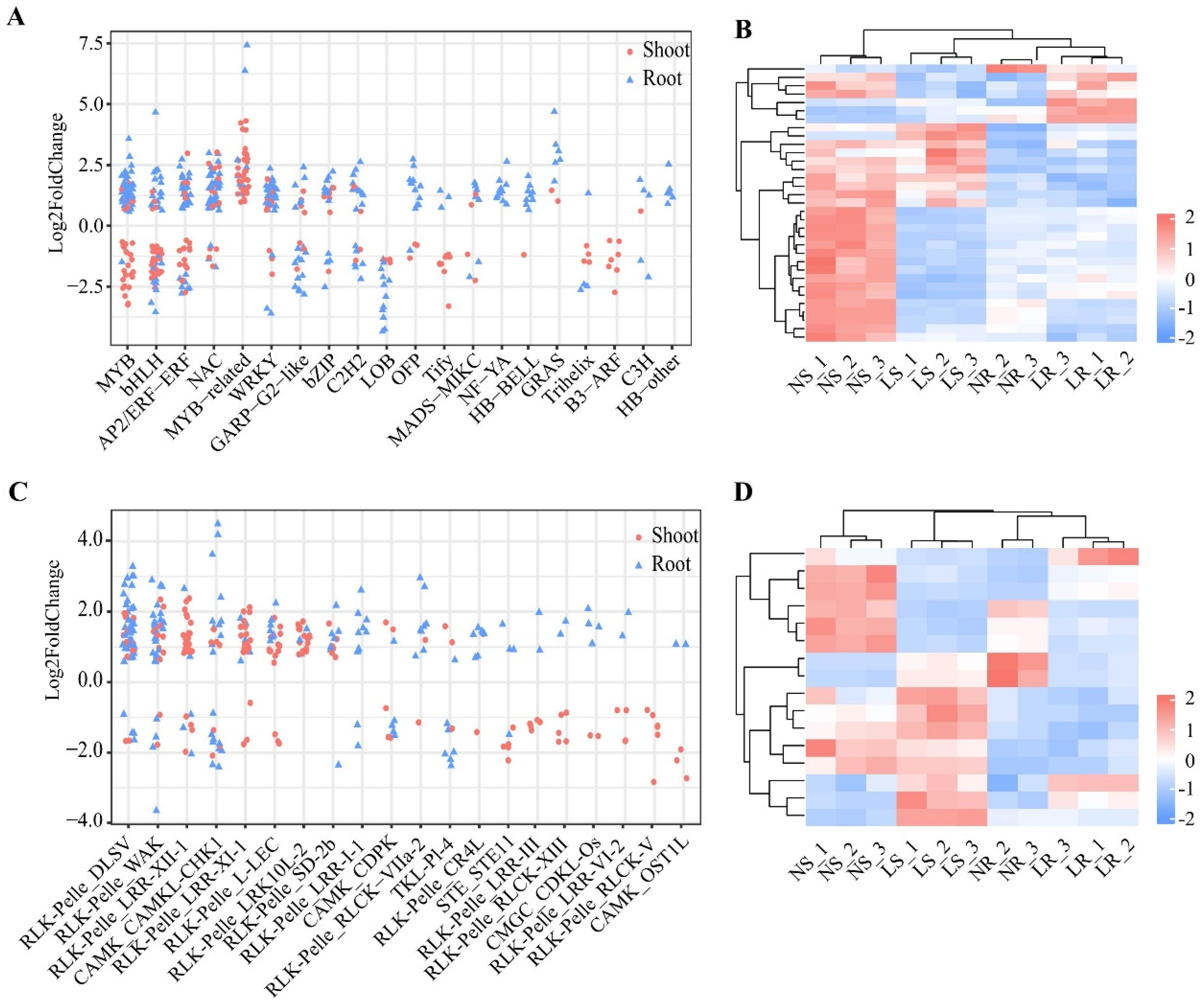
2.5. Identification of Differentially Expressed Transcription Factors and Protein Kinases
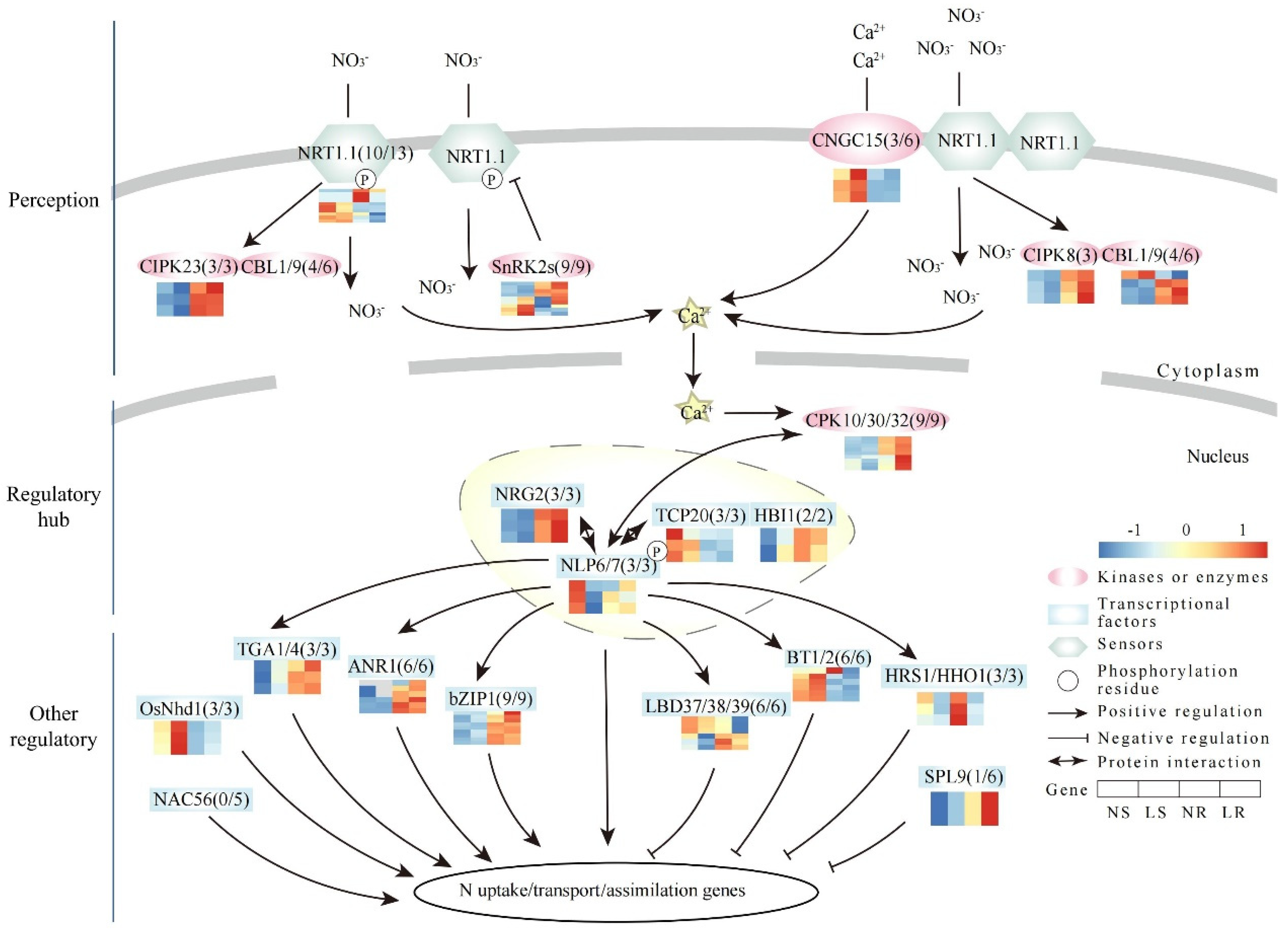
2.6. Expression Pattern of Nitrate Signaling Pathway Genes
2.7. Identification of Co-Expression Genes in Response to LN Stress
3. Discussion
3.1. LN Stress Response in Wheat
3.2. Nitrate Signaling Network in Wheat
4. Materials and Methods
4.1. Plant Materials and Treatment
4.2. RNA Isolation, Library Construction and Sequencing
4.3. Read Preprocessing and Identification of DEGs
4.4. Function Annotation
4.5. Identification of Orthologous Genes
4.6. Weighted Gene Co-Expression Network Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Juliana, P.; Poland, J.; Huerta-Espino, J.; Shrestha, S.; Crossa, J.; Crespo-Herrera, L.; Toledo, F.H.; Govindan, V.; Mondal, S.; Kumar, U.; et al. Improving grain yield, stress resilience and quality of bread wheat using large-scale genomics. Nat. Genet. 2019, 51, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Baer-Nawrocka, A.; Sadowski, A. Food security and food self-sufficiency around the world: A typology of countries. PLoS ONE 2019, 14, e0213448. [Google Scholar] [CrossRef] [PubMed]
- Bloch, S.E.; Ryu, M.H.; Ozaydin, B.; Broglie, R. Harnessing atmospheric nitrogen for cereal crop production. Curr. Opin. Biotechnol. 2020, 62, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, L.; Bai, P.; Wei, K.; Zhang, Y.; Ruan, L.; Wu, L.; Cheng, H. Identification of Regulatory Networks and Hub Genes Controlling Nitrogen Uptake in Tea Plants [Camellia sinensis (L.) O. Kuntze]. J. Agric. Food Chem. 2020, 68, 2445–2456. [Google Scholar] [CrossRef] [PubMed]
- Zörb, C.; Ludewig, U.; Hawkesford, M.J. Perspective on Wheat Yield and Quality with Reduced Nitrogen Supply. Trends Plant Sci. 2018, 23, 1029–1037. [Google Scholar] [CrossRef]
- Barbieri, P.; Pellerin, S.; Seufert, V.; Smith, L.; Ramankutty, N.; Nesme, T. Global option space for organic agriculture is delimited by nitrogen availability. Nat. Food 2021, 2, 363–372. [Google Scholar] [CrossRef]
- Matassa, S.; Boeckx, P.; Boere, J.; Erisman, J.W.; Guo, M.; Manzo, R.; Meerburg, F.; Papirio, S.; Pikaar, I.; Rabaey, K.; et al. How can we possibly resolve the planet’s nitrogen dilemma? Microb. Biotechnol. 2023, 16, 15–27. [Google Scholar] [CrossRef]
- Tang, W.; He, X.; Qian, L.; Wang, F.; Zhang, Z.; Sun, C.; Lin, L.; Guan, C. Comparative Transcriptome Analysis in Oilseed Rape (Brassica napus) Reveals Distinct Gene Expression Details between Nitrate and Ammonium Nutrition. Genes 2019, 10, 391. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Cheng, Y.H.; Chen, K.E.; Tsay, Y.F. Nitrate Transport, Signaling, and Use Efficiency. Annu. Rev. Plant Biol. 2018, 69, 85–122. [Google Scholar] [CrossRef]
- Vidal, E.A.; Alvarez, J.M.; Araus, V.; Riveras, E.; Brooks, M.D.; Krouk, G.; Ruffel, S.; Lejay, L.; Crawford, N.M.; Coruzzi, G.M.; et al. Nitrate in 2020: Thirty Years from Transport to Signaling Networks. Plant Cell 2020, 32, 2094–2119. [Google Scholar] [CrossRef]
- Chaput, V.; Li, J.; Séré, D.; Tillard, P.; Fizames, C.; Moyano, T.; Zuo, K.; Martin, A.; Gutiérrez, R.A.; Gojon, A.; et al. Characterization of the signalling pathways involved in the repression of root nitrate uptake by nitrate in Arabidopsis thaliana. J. Exp. Bot. 2023, 74, 4244–4258. [Google Scholar] [CrossRef]
- Zhang, Z.; Hu, B.; Chu, C. Towards understanding the hierarchical nitrogen signalling network in plants. Curr. Opin. Plant Biol. 2020, 55, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Kant, S. Understanding nitrate uptake, signaling and remobilisation for improving plant nitrogen use efficiency. Semin. Cell Dev. Biol. 2018, 74, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.C.; Chiang, C.S.; Crawford, N.M.; Tsay, Y.F. CHL1 encodes a component of the low-affinity nitrate uptake system in Arabidopsis and shows cell type-specific expression in roots. Plant Cell 1996, 8, 2183–2191. [Google Scholar] [CrossRef]
- Maghiaoui, A.; Gojon, A.; Bach, L. NRT1.1-centered nitrate signaling in plants. J. Exp. Bot. 2020, 71, 6226–6237. [Google Scholar] [CrossRef] [PubMed]
- Riveras, E.; Alvarez, J.M.; Vidal, E.A.; Oses, C.; Vega, A.; Gutiérrez, R.A. The Calcium Ion Is a Second Messenger in the Nitrate Signaling Pathway of Arabidopsis. Plant Physiol. 2015, 169, 1397–1404. [Google Scholar] [CrossRef]
- Su, H.; Wang, T.; Ju, C.; Deng, J.; Zhang, T.; Li, M.; Tian, H.; Wang, C. Abscisic acid signaling negatively regulates nitrate uptake via phosphorylation of NRT1.1 by SnRK2s in Arabidopsis. J. Integr. Plant Biol. 2021, 63, 597–610. [Google Scholar] [CrossRef]
- Liu, L.; Gao, H.; Li, S.; Han, Z.; Li, B. Calcium signaling networks mediate nitrate sensing and responses in Arabidopsis. Plant Signal. Behav. 2021, 16, 1938441. [Google Scholar] [CrossRef]
- Liu, K.H.; Niu, Y.; Konishi, M.; Wu, Y.; Du, H.; Sun Chung, H.; Li, L.; Boudsocq, M.; McCormack, M.; Maekawa, S.; et al. Discovery of nitrate-CPK-NLP signalling in central nutrient-growth networks. Nature 2017, 545, 311–316. [Google Scholar] [CrossRef]
- Zhang, T.T.; Kang, H.; Fu, L.L.; Sun, W.J.; Gao, W.S.; You, C.X.; Wang, X.F.; Hao, Y.J. NIN-like protein 7 promotes nitrate-mediated lateral root development by activating transcription of TRYPTOPHAN AMINOTRANSFERASE RELATED 2. Plant Sci. Int. J. Exp. Plant Biol. 2021, 303, 110771. [Google Scholar] [CrossRef]
- Konishi, M.; Okitsu, T.; Yanagisawa, S. Nitrate-responsive NIN-like protein transcription factors perform unique and redundant roles in Arabidopsis. J. Exp. Bot. 2021, 72, 5735–5750. [Google Scholar] [CrossRef]
- Xu, N.; Wang, R.; Zhao, L.; Zhang, C.; Li, Z.; Lei, Z.; Liu, F.; Guan, P.; Chu, Z.; Crawford, N.M.; et al. The Arabidopsis NRG2 Protein Mediates Nitrate Signaling and Interacts with and Regulates Key Nitrate Regulators. Plant Cell 2016, 28, 485–504. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.; Wang, R.; Nacry, P.; Breton, G.; Kay, S.A.; Pruneda-Paz, J.L.; Davani, A.; Crawford, N.M. Nitrate foraging by Arabidopsis roots is mediated by the transcription factor TCP20 through the systemic signaling pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 15267–15272. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Li, M.; Zhang, S.; Fan, M.; Han, C.; Xiang, F.; Li, G.; Wang, Y.; Xiang, C.B.; Wang, J.G.; et al. HBI1-TCP20 interaction positively regulates the CEPs-mediated systemic nitrate acquisition. J. Integr. Plant Biol. 2021, 63, 902–912. [Google Scholar] [CrossRef]
- Canales, J.; Contreras-López, O.; Álvarez, J.M.; Gutiérrez, R.A. Nitrate induction of root hair density is mediated by TGA1/TGA4 and CPC transcription factors in Arabidopsis thaliana. Plant J. Cell Mol. Biol. 2017, 92, 305–316. [Google Scholar] [CrossRef]
- Obertello, M.; Krouk, G.; Katari, M.S.; Runko, S.J.; Coruzzi, G.M. Modeling the global effect of the basic-leucine zipper transcription factor 1 (bZIP1) on nitrogen and light regulation in Arabidopsis. BMC Syst. Biol. 2010, 4, 111. [Google Scholar] [CrossRef]
- Para, A.; Li, Y.; Marshall-Colón, A.; Varala, K.; Francoeur, N.J.; Moran, T.M.; Edwards, M.B.; Hackley, C.; Bargmann, B.O.; Birnbaum, K.D.; et al. Hit-and-run transcriptional control by bZIP1 mediates rapid nutrient signaling in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, 10371–10376. [Google Scholar] [CrossRef]
- Xu, P.; Ma, W.; Hu, J.; Cai, W. The nitrate-inducible NAC transcription factor NAC056 controls nitrate assimilation and promotes lateral root growth in Arabidopsis thaliana. PLoS Genet. 2022, 18, e1010090. [Google Scholar] [CrossRef]
- Li, K.; Zhang, S.; Tang, S.; Zhang, J.; Dong, H.; Yang, S.; Qu, H.; Xuan, W.; Gu, M.; Xu, G. The rice transcription factor Nhd1 regulates root growth and nitrogen uptake by activating nitrogen transporters. Plant Physiol. 2022, 189, 1608–1624. [Google Scholar] [CrossRef]
- Zhang, H.; Forde, B.G. An Arabidopsis MADS box gene that controls nutrient-induced changes in root architecture. Science 1998, 279, 407–409. [Google Scholar] [CrossRef]
- Kiba, T.; Inaba, J.; Kudo, T.; Ueda, N.; Konishi, M.; Mitsuda, N.; Takiguchi, Y.; Kondou, Y.; Yoshizumi, T.; Ohme-Takagi, M.; et al. Repression of Nitrogen Starvation Responses by Members of the Arabidopsis GARP-Type Transcription Factor NIGT1/HRS1 Subfamily. Plant Cell 2018, 30, 925–945. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, L.; Li, Y.; Zhang, D.; Gao, Y. Plant NIGT1/HRS1/HHO Transcription Factors: Key Regulators with Multiple Roles in Plant Growth, Development, and Stress Responses. Int. J. Mol. Sci. 2021, 22, 8685. [Google Scholar] [CrossRef] [PubMed]
- Medici, A.; Marshall-Colon, A.; Ronzier, E.; Szponarski, W.; Wang, R.; Gojon, A.; Crawford, N.M.; Ruffel, S.; Coruzzi, G.M.; Krouk, G. AtNIGT1/HRS1 integrates nitrate and phosphate signals at the Arabidopsis root tip. Nat. Commun. 2015, 6, 6274. [Google Scholar] [CrossRef] [PubMed]
- Rubin, G.; Tohge, T.; Matsuda, F.; Saito, K.; Scheible, W.R. Members of the LBD family of transcription factors repress anthocyanin synthesis and affect additional nitrogen responses in Arabidopsis. Plant Cell 2009, 21, 3567–3584. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.; Chen, J.; Yue, X.; Li, C.; Zhang, Q.; de Dios, V.R.; Yao, Y.; Tan, W. Interaction of BES1 and LBD37 transcription factors modulates brassinosteroid-regulated root forging response under low nitrogen in arabidopsis. Front. Plant Sci. 2022, 13, 998961. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Maekawa, S.; Konishi, M.; Yoshioka, N.; Sasaki, Y.; Maeda, H.; Ishida, T.; Kato, Y.; Yamaguchi, J.; Yanagisawa, S. Direct transcriptional activation of BT genes by NLP transcription factors is a key component of the nitrate response in Arabidopsis. Biochem. Biophys. Res. Commun. 2017, 483, 380–386. [Google Scholar] [CrossRef]
- Krouk, G.; Mirowski, P.; LeCun, Y.; Shasha, D.E.; Coruzzi, G.M. Predictive network modeling of the high-resolution dynamic plant transcriptome in response to nitrate. Genome Biol. 2010, 11, R123. [Google Scholar] [CrossRef]
- Bayega, A.; Fahiminiya, S.; Oikonomopoulos, S.; Ragoussis, J. Current and Future Methods for mRNA Analysis: A Drive Toward Single Molecule Sequencing. Methods Mol. Biol. 2018, 1783, 209–241. [Google Scholar] [CrossRef]
- Zhang, H.; He, L.; Cai, L. Transcriptome Sequencing: RNA-Seq. Methods Mol. Biol. 2018, 1754, 15–27. [Google Scholar] [CrossRef]
- Xin, W.; Zhang, L.; Zhang, W.; Gao, J.; Yi, J.; Zhen, X.; Du, M.; Zhao, Y.; Chen, L. An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Root Growth Regulation Mechanisms in Response to Nitrogen Availability. Int. J. Mol. Sci. 2019, 20, 5893. [Google Scholar] [CrossRef]
- Quan, X.; Zeng, J.; Chen, G.; Zhang, G. Transcriptomic analysis reveals adaptive strategies to chronic low nitrogen in Tibetan wild barley. BMC Plant Biol. 2019, 19, 68. [Google Scholar] [CrossRef]
- Krapp, A.; Berthomé, R.; Orsel, M.; Mercey-Boutet, S.; Yu, A.; Castaings, L.; Elftieh, S.; Major, H.; Renou, J.P.; Daniel-Vedele, F. Arabidopsis roots and shoots show distinct temporal adaptation patterns toward nitrogen starvation. Plant Physiol. 2011, 157, 1255–1282. [Google Scholar] [CrossRef] [PubMed]
- Joshi, V.; Joshi, M.; Penalosa, A. Comparative analysis of tissue-specific transcriptomic responses to nitrogen stress in spinach (Spinacia oleracea). PLoS ONE 2020, 15, e0232011. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, M.A.; Chen, C.; Shireen, F.; Zheng, Z.; Sohail, H.; Afzal, M.; Ali, M.A.; Bie, Z.; Huang, Y. Genome-wide expression profiling of leaves and roots of watermelon in response to low nitrogen. BMC Genom. 2018, 19, 456. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Du, R.; Li, Z.; Chen, Z.; Li, J.; Du, H. An integrated nitrogen utilization gene network and transcriptome analysis reveal candidate genes in response to nitrogen deficiency in Brassica napus. Front. Plant Sci. 2023, 14, 1187552. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Zhao, Y.; Zhang, Y.; Zhang, J.; Ma, H.; Han, Y. Transcriptome analysis reveals Nitrogen deficiency induced alterations in leaf and root of three cultivars of potato (Solanum tuberosum L.). PLoS ONE 2020, 15, e0240662. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.K.; Sevanthi, V.A.; Chaudhary, S.; Tyagi, P.; Venkadesan, S.; Rani, M.; Mandal, P.K. Transcriptome Analysis of Two Rice Varieties Contrasting for Nitrogen Use Efficiency under Chronic N Starvation Reveals Differences in Chloroplast and Starch Metabolism-Related Genes. Genes 2018, 9, 206. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Q.; Zhang, S.; Wu, P.; Chen, Y.; Li, M.; Jiang, H.; Wu, G. Global gene expression analysis of the response of physic nut (Jatropha curcas L.) to medium- and long-term nitrogen deficiency. PLoS ONE 2017, 12, e0182700. [Google Scholar] [CrossRef]
- Schlüter, U.; Mascher, M.; Colmsee, C.; Scholz, U.; Bräutigam, A.; Fahnenstich, H.; Sonnewald, U. Maize source leaf adaptation to nitrogen deficiency affects not only nitrogen and carbon metabolism but also control of phosphate homeostasis. Plant Physiol. 2012, 160, 1384–1406. [Google Scholar] [CrossRef]
- Mascia, M.; Sega, D.; Zamboni, A.; Varanini, Z. Nitrogen Starvation Differentially Influences Transcriptional and Uptake Rate Profiles in Roots of Two Maize Inbred Lines with Different NUE. Int. J. Mol. Sci. 2019, 20, 4856. [Google Scholar] [CrossRef]
- Wang, J.; Song, K.; Sun, L.; Qin, Q.; Sun, Y.; Pan, J.; Xue, Y. Morphological and Transcriptome Analysis of Wheat Seedlings Response to Low Nitrogen Stress. Plants 2019, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Shamshad, M.; Jindal, S.; Kaur, A.; Singh, S.; Sharma, A.; Kaur, S. RNA-Seq-Based Transcriptomics Study to Investigate the Genes Governing Nitrogen Use Efficiency in Indian Wheat Cultivars. Front. Genet. 2022, 13, 853910. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yin, C.; Xiang, L.; Jiang, W.; Xu, S.; Mao, Z. Transcription strategies related to photosynthesis and nitrogen metabolism of wheat in response to nitrogen deficiency. BMC Plant Biol. 2020, 20, 448. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Shi, H.; Hu, C.; Luo, M.; Xu, C.; Wang, S.; Li, N.; Tang, W.; Zhou, Y.; Wang, C.; et al. Transcriptome Differences in Response Mechanisms to Low-Nitrogen Stress in Two Wheat Varieties. Int. J. Mol. Sci. 2021, 22, 12278. [Google Scholar] [CrossRef]
- Curci, P.L.; Aiese Cigliano, R.; Zuluaga, D.L.; Janni, M.; Sanseverino, W.; Sonnante, G. Transcriptomic response of durum wheat to nitrogen starvation. Sci. Rep. 2017, 7, 1176. [Google Scholar] [CrossRef]
- Wang, H.; Ma, Q.; Shan, F.; Tian, L.; Gong, J.; Quan, W.; Yang, W.; Hou, Q.; Zhang, F.; Zhang, S. Transcriptional regulation mechanism of wheat varieties with different nitrogen use efficiencies in response to nitrogen deficiency stress. BMC Genom. 2022, 23, 727. [Google Scholar] [CrossRef]
- Beleggia, R.; Omranian, N.; Holtz, Y.; Gioia, T.; Fiorani, F.; Nigro, F.M.; Pecchioni, N.; De Vita, P.; Schurr, U.; David, J.L.; et al. Comparative Analysis Based on Transcriptomics and Metabolomics Data Reveal Differences between Emmer and Durum Wheat in Response to Nitrogen Starvation. Int. J. Mol. Sci. 2021, 22, 4790. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Du, W.; Xia, H.; Fan, S.; Zhang, B. Signaling Responses to N Starvation: Focusing on Wheat and Filling the Putative Gaps With Findings Obtained in Other Plants. A Review. Front. Plant Sci. 2021, 12, 656696. [Google Scholar] [CrossRef]
- Gong, Z.; Xiong, L.; Shi, H.; Yang, S.; Herrera-Estrella, L.R.; Xu, G.; Chao, D.Y.; Li, J.; Wang, P.Y.; Qin, F.; et al. Plant abiotic stress response and nutrient use efficiency. Sci. China Life Sci. 2020, 63, 635–674. [Google Scholar] [CrossRef]
- Qiao, L.; Zhang, L.; Zhang, X.; Zhang, L.; Li, X.; Chang, J.; Zhan, H.; Guo, H.; Zheng, J.; Chang, Z. Evolution of the Aux/IAA Gene Family in Hexaploid Wheat. J. Mol. Evol. 2017, 85, 107–119. [Google Scholar] [CrossRef]
- Hu, B.; Wang, W.; Ou, S.; Tang, J.; Li, H.; Che, R.; Zhang, Z.; Chai, X.; Wang, H.; Wang, Y.; et al. Variation in NRT1.1B contributes to nitrate-use divergence between rice subspecies. Nat. Genet. 2015, 47, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Feng, H.; Tan, Y.; Xu, Y.; Miao, Q.; Xu, G. A putative 6-transmembrane nitrate transporter OsNRT1.1b plays a key role in rice under low nitrogen. J. Integr. Plant Biol. 2016, 58, 590–599. [Google Scholar] [CrossRef]
- Jagadhesan, B.; Sathee, L.; Meena, H.S.; Jha, S.K.; Chinnusamy, V.; Kumar, A.; Kumar, S. Genome wide analysis of NLP transcription factors reveals their role in nitrogen stress tolerance of rice. Sci. Rep. 2020, 10, 9368. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Batra, R.; Gahlaut, V.; Gautam, T.; Kumar, S.; Sharma, M.; Tyagi, S.; Singh, K.P.; Balyan, H.S.; Pandey, R.; et al. Genome-wide identification and characterization of gene family for RWP-RK transcription factors in wheat (Triticum aestivum L.). PLoS ONE 2018, 13, e0208409. [Google Scholar] [CrossRef]
- Sawaki, N.; Tsujimoto, R.; Shigyo, M.; Konishi, M.; Toki, S.; Fujiwara, T.; Yanagisawa, S. A nitrate-inducible GARP family gene encodes an auto-repressible transcriptional repressor in rice. Plant Cell Physiol. 2013, 54, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. Publ. Protein Soc. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2023, 51, D418–D427. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Zhang, X.; Li, F.; Ding, Y.; Ma, Q.; Yi, Y.; Zhu, M.; Ding, J.; Li, C.; Guo, W.; Zhu, X. Transcriptome Analysis of Two Near-Isogenic Lines with Different NUE under Normal Nitrogen Conditions in Wheat. Biology 2021, 10, 787. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Libraries | Row Reads | Clean Reads | GC (%) | Error Rate (%) | Q20 (%) | Q30 (%) | Mapping Alignment Rate (%) | Uniq Mapped Reads (%) | Multi Mapped Reads (%) |
|---|---|---|---|---|---|---|---|---|---|
| LS_1 | 52,362,666 | 49,208,442 | 56.07 | 0.02 | 98.06 | 94.49 | 98.63 | 90.87 | 7.76 |
| LS_2 | 57,022,090 | 54,302,414 | 56.97 | 0.03 | 97.83 | 93.99 | 98.56 | 91.27 | 7.29 |
| LS_3 | 45,084,032 | 41,501,168 | 53.32 | 0.02 | 98 | 94.36 | 98.34 | 87.72 | 10.62 |
| LR_1 | 46,019,256 | 44,010,914 | 54.31 | 0.03 | 97.8 | 93.93 | 97.85 | 91.23 | 6.61 |
| LR_2 | 43,574,686 | 41,582,874 | 54.66 | 0.03 | 97.89 | 94.17 | 97.75 | 91.13 | 6.62 |
| LR_3 | 43,976,886 | 41,912,238 | 53.93 | 0.03 | 97.77 | 93.8 | 97.74 | 90.83 | 6.91 |
| NS_1 | 56,016,618 | 53,647,952 | 58.09 | 0.03 | 97.74 | 93.81 | 98.61 | 90.39 | 8.23 |
| NS_2 | 46,546,442 | 44,198,178 | 55.93 | 0.03 | 97.88 | 94.14 | 98.61 | 89.45 | 9.17 |
| NS_3 | 49,068,108 | 46,009,472 | 55.32 | 0.02 | 97.96 | 94.3 | 98.43 | 81.45 | 16.99 |
| NR_2 | 49,085,898 | 46,338,388 | 53.65 | 0.02 | 97.97 | 94.3 | 97.24 | 90.09 | 7.15 |
| NR_3 | 47,677,344 | 44,892,372 | 53.08 | 0.03 | 97.32 | 92.35 | 97.18 | 89.83 | 7.35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, P.; Zhu, Y.; Shang, Y.; Wu, Z.; Tao, Y.; Wang, H.; Li, D.; Zhang, C. Transcriptome Profiling Reveals the Gene Network Responding to Low Nitrogen Stress in Wheat. Plants 2024, 13, 371. https://doi.org/10.3390/plants13030371
Wang Y, Li P, Zhu Y, Shang Y, Wu Z, Tao Y, Wang H, Li D, Zhang C. Transcriptome Profiling Reveals the Gene Network Responding to Low Nitrogen Stress in Wheat. Plants. 2024; 13(3):371. https://doi.org/10.3390/plants13030371
Chicago/Turabian StyleWang, Yiwei, Pengfeng Li, Yiwang Zhu, Yuping Shang, Zhiqiang Wu, Yongfu Tao, Hongru Wang, Dongxi Li, and Cuijun Zhang. 2024. "Transcriptome Profiling Reveals the Gene Network Responding to Low Nitrogen Stress in Wheat" Plants 13, no. 3: 371. https://doi.org/10.3390/plants13030371
APA StyleWang, Y., Li, P., Zhu, Y., Shang, Y., Wu, Z., Tao, Y., Wang, H., Li, D., & Zhang, C. (2024). Transcriptome Profiling Reveals the Gene Network Responding to Low Nitrogen Stress in Wheat. Plants, 13(3), 371. https://doi.org/10.3390/plants13030371