1. Introduction
Oil tea camellia (
Camellia spp.), also known as “Shanyou”, is a perennial evergreen shrub or small tree that belongs to the family Theaceae and genus
Camellia L. It is a woody oil crop famous for the high oil content of its seeds [
1,
2]. This plant has a long history of cultivation in Eastern and Southern Asia, particularly in China, and is an important woody oil cash crop. Oil tea camellia is recognized globally as one of the four major woody oilseed crops together with oil palm (
Elaeis guineensis Jacq.), olive (
Olea europaea L.), and coconut (
Cocos nucifera Linn.). Its advantages include a strong adaptability, drought resistance, and infertile soil resistance [
3]. There remains limited research on the specific genetic diversity differences between octoploid and decaploid populations in tropical environments. This study aims to fill this gap by providing insights into how ploidy levels and geographic factors contribute to genetic variation, particularly in the underexplored tropical regions of Hainan. Because of its high economic value, the extensive scope of comprehensive development, a diverse range of applications, and significant utilization potential, it is considered to be a “guaranteed high-yield crop” and “green oil bank” by farmers [
4].
Oil tea camellia is primarily distributed in the Yangtze River Basin and southern China but is also grown in small quantities in countries such as Myanmar, Thailand, and Vietnam. In China, oil tea camellia is mainly cultivated in the Hunan, Jiangxi, and Guangxi provinces, with these three regions together accounting for 75.8% of China’s total oil tea camellia cultivation area, with Hunan in particular exhibiting the widest cultivation area accounting for approximately 40% of the total [
5].
Camellia vietnamensis is particularly suitable for cultivation at low altitudes in the tropical regions of Southern Asia because of its fast growth rate to tall heights and its adaptability to the tropical climate [
6]. In the Leizhou Peninsula of the Guangdong Province and from the southeastern part of the Guangxi Zhuang Autonomous Region to Hainan Province,
Camellia vietnamensis has adapted to climatic conditions and exhibits good flowering and fruiting characteristics.
Camellia vietnamensis cultivated in Hainan is thought to have originated in Gaozhou City, Guangdong Province; therefore, it is also known as “
Camellia gauchowensis Chang” in the Hainan region. The history of oil tea camellia cultivation in Hainan may be traced back more than 500 years [
7]. A systematic study of the resource distribution of oil tea camellia in Hainan and a field survey revealed that the oil tea camellia germplasm resources in Hainan include wild and artificial cultivars, which are primarily concentrated in nine county-level cities and thirty-eight townships, encompassing approximately 1167.3 hectares [
8].
Hainan Island is rich in oil tea camellia resources. The book
Flora of China indicates that common native oil tea camellia species in Hainan grow in old-growth forests at altitudes above 800 m. According to the
Flora Hainanica,
A Checklist of Flowering Plants of Islands and Reefs of Hainan and Guangdong Province,
Hainan Island Crop (Plant) Germplasm Resources Investigation Anthology, and other sources, Hainan Island contains a wealth of wild common oil tea camellia germplasm resources. Considerable evidence indicates that Hainan Island is the origin of the tropical oil tea camellia and has a long history of oil tea camellia cultivation. In numerous cities and counties in Hainan Province, there exist many oil tea camellia trees that are more than 100 years old, or even several 100 years old, particularly in Ding’an, Chengmai, Tunchang, and Qionghai, where oil tea camellia forests of an older age are preserved. Chengmai County has the highest concentration of oil tea camellia forests, with the largest trees having basal diameters up to 150 cm [
8].
Whole-genome sequencing (WGS) is a precise technology that comprehensively reveals an organism’s genetic information. This enables researchers to gain insight into its genetic background and biological functions. Whole genome resequencing (WGR), meanwhile, refers to the sequencing of the genome of a specific individual or population based on an existing reference genome to discover genome-wide variations, such as single nucleotide polymorphisms (SNPs), structural variants, insertion/deletion variation sites (InDels), and copy number variations. These can be used to analyze the molecular genetic characteristics of an individual or population, screen and predict the genes for key economic traits, and study genetic evolution [
9].
In January 2022, a research team from the Research Institute of Sub-Forestry, Chinese Academy of Forestry, used diploid oil tea camellia to successfully map the entire genome of oil tea camellia, the results of which clearly illustrated the origin and evolution of oil tea camellia. After more than 4 years of continuous study, this research team successfully obtained a diploid oil tea camellia genome map with a size of approximately 2.95 GB and a Contig N50 of 1.002 MB by applying PacBio third-generation sequencing technology. They accurately localized the genome sequences to 15 chromosomes, achieving a high localization rate of 91.33% [
10]. This represents the first high-quality oil tea camellia genome map with chromosome-level precision, setting an example for the assembly of a complex genome of a woody oilseed crop. As of 2023, the whole genomes of some species within the genus
Camellia of the family Theaceae had been sequenced. Besides oil tea camellia,
Camellia sinensis,
Camellia japonica,
Camellia nitidissima,
Camellia sinensis var.
pubilimba, and
Stewartia sinensis have been sequenced. This is a relatively small number compared with the families and genera of other crops.
At present, some progress has been made in the research of oil tea camellia WGR, which has provided a foundation for evaluating and utilizing oil tea camellia germplasm resources, molecular selective breeding, and the selection of high-quality varieties. oil tea camellia is widely distributed in China, and its genetic diversity is rich across these different geographic environments. Hainan oil tea camellia and that of the inland exhibit different genetic backgrounds and adaptive characteristics due to differences in their geographic location and climate. These differences are likely to be closely related to variations in ploidy level and genome structure. In this study, we selected 49 oil tea camellia germplasm resources from five tropical oil tea camellia populations in Hainan, Guangdong, and Guangxi provinces. After determining the ploidy level of the samples through flow cytometry, we performed WGR analysis to obtain the SNP and InDel variation data from the oil tea camellia samples. Additionally, we used bioinformatics to explore the differences and genetic diversity between octoploid and decaploid oil tea camellia in the tropics and between oil tea camellia in Hainan and the inland. Our findings provide a reference for future studies of oil tea camellia selective breeding and the conservation, rational use, and genetic modification of germplasm resources.
3. Discussion
WGR technology may be used to obtain information on genome-wide locus variation. It has been widely used in gene localization, genetic map construction, mutation site identification, and genetic evolution research [
9,
11]. Genetic diversity plays a crucial role in the evolutionary capacity of species; those with a higher genetic diversity tend to adapt more efficiently to changes in the environment, whereas those with a lower genetic diversity may gradually lose their competitiveness and become less adaptable through long-term natural selection [
12].
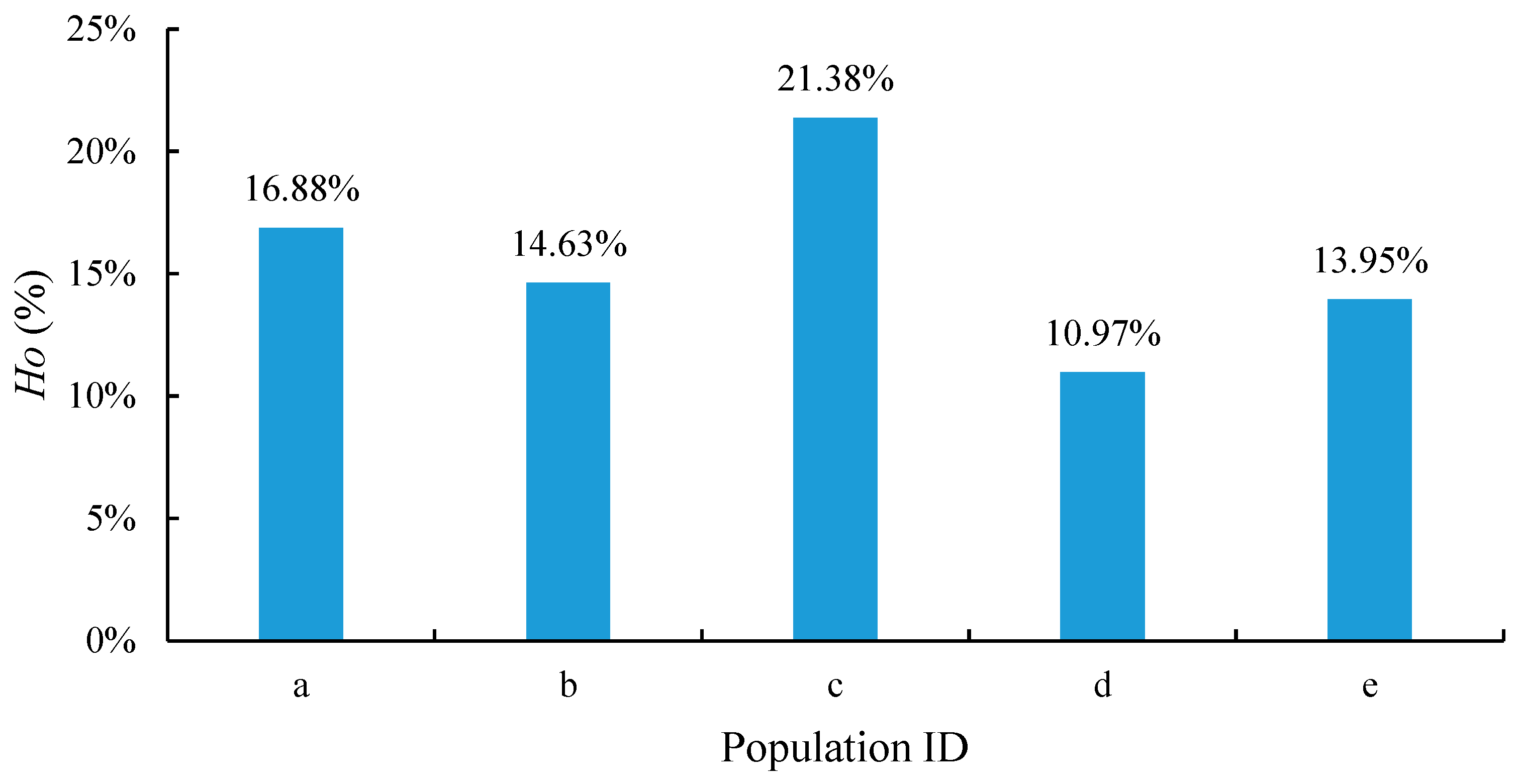
In the present study, we used genome-wide allele frequencies, the heterozygosity (
Ho) of oil tea camellia, and the diversity indices of phenotypic traits to assess genetic diversity. A high heterozygosity is usually associated with a good adaptive capacity and survival potential. The results indicated the presence of a large number of low-frequency alleles in the sample genomes, which suggests a high genetic diversity and extensive genetic variation in the oil tea camellia populations. Of the five oil tea camellia populations in the sample genome, population c achieved the largest
Ho value, indicating a higher genetic diversity among this population compared with the others. Populations a and b were more genetically diverse than d and e. The high genetic diversity observed in oil tea camellia populations is consistent with the findings of Ye et al. [
13], who highlighted significant genetic diversity across various Camellia populations, partly attributing this diversity to environmental adaptation and ploidy-level variation. This rich genetic diversity provides a foundation for breeding programs targeting traits like ecological adaptation, oil yield, genetic modification, and biodiversity conservation of the species.
In addition to its role in breeding and adaptation, maintaining a high genetic diversity is crucial for preserving the beneficial bioactive compounds present in oil tea camellia, which are valuable for medicinal and commercial applications. Teixeira and Sousa [
14] emphasized the diverse biological activities of Camellia species, including their antioxidant, anti-inflammatory, and anti-cancer properties, which are linked to the genetic variability of these plants. Additionally, Li et al. [
15] further elaborated on the therapeutic benefits of camellia oil, highlighting its positive effects on cardiovascular health due to its rich unsaturated fatty acid content. Therefore, the diversity observed in our study is instrumental in enhancing these health-promoting compounds, and conservation efforts should focus on maintaining this genetic diversity to maximize both the medicinal and economic value of oil tea camellia.
The above findings not only enhance our understanding of the oil tea camellia genome, but also provide a basis for the conservation of its germplasm resources and genetic modification. However, it is important to consider ethical issues related to genetic modification. While our study provides a foundation for potential genetic modifications, any practical applications should undergo careful evaluation regarding their ecological impacts and ethical implications. Future studies may further explore how ploidy-level and environmental factors specifically affect the genetic structure and gene expression of oil tea camellia and how these differences influence the survival and reproduction strategies and the evolutionary direction of oil tea camellia. However, some of the observed differences could not be explained in this study because of the large number of genomes associated with the octoploid and decaploid oil tea camellia samples. This was also affected by the presence of a large number of DNA repeated sequences, high heterozygosity, genome complexity, and detection errors in the sequencing methods and technology. In the future, sequencing methodology and technology improvements are needed to further reduce detection errors.
The WGR study has increased our understanding of the genetic diversity of oil tea camellia through variation detection and population genetic analysis. Our findings provide strong support for the discovery of high-quality alleles and the genetic analysis of important agronomic traits. Additionally, the resequencing data may be used to identify various molecular markers, construct high-density genetic maps, and accelerate the molecular breeding process of oil tea camellia. However, there are still important issues to be solved in terms of the WGR of oil tea camellia, such as the relatively high sequencing cost and major challenges such as the huge amount of genome data, the numerous repeated sequences, and the extremely complex homologous and heterologous structures of the various subgenomes of oil tea camellia. Additionally, data analysis and bioinformatics processing also pose challenges, and the validation of and functional research into the various sites must be strengthened. With the increasing maturity of sequencing technology and the continuous improvement of bioinformatics analysis, more breakthroughs are bound to be achieved in future research on the WGR of oil tea camellia.
5. Conclusions
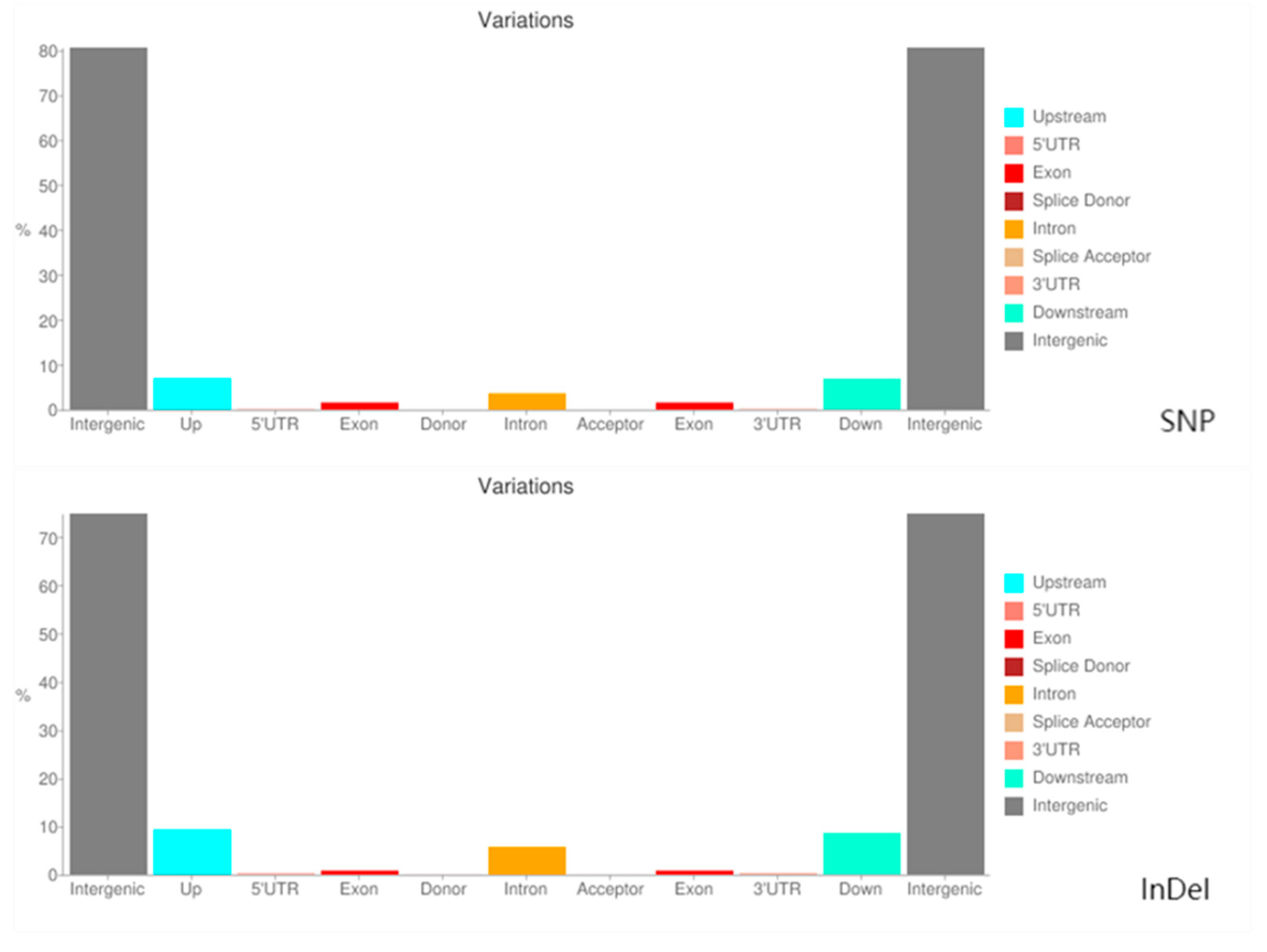
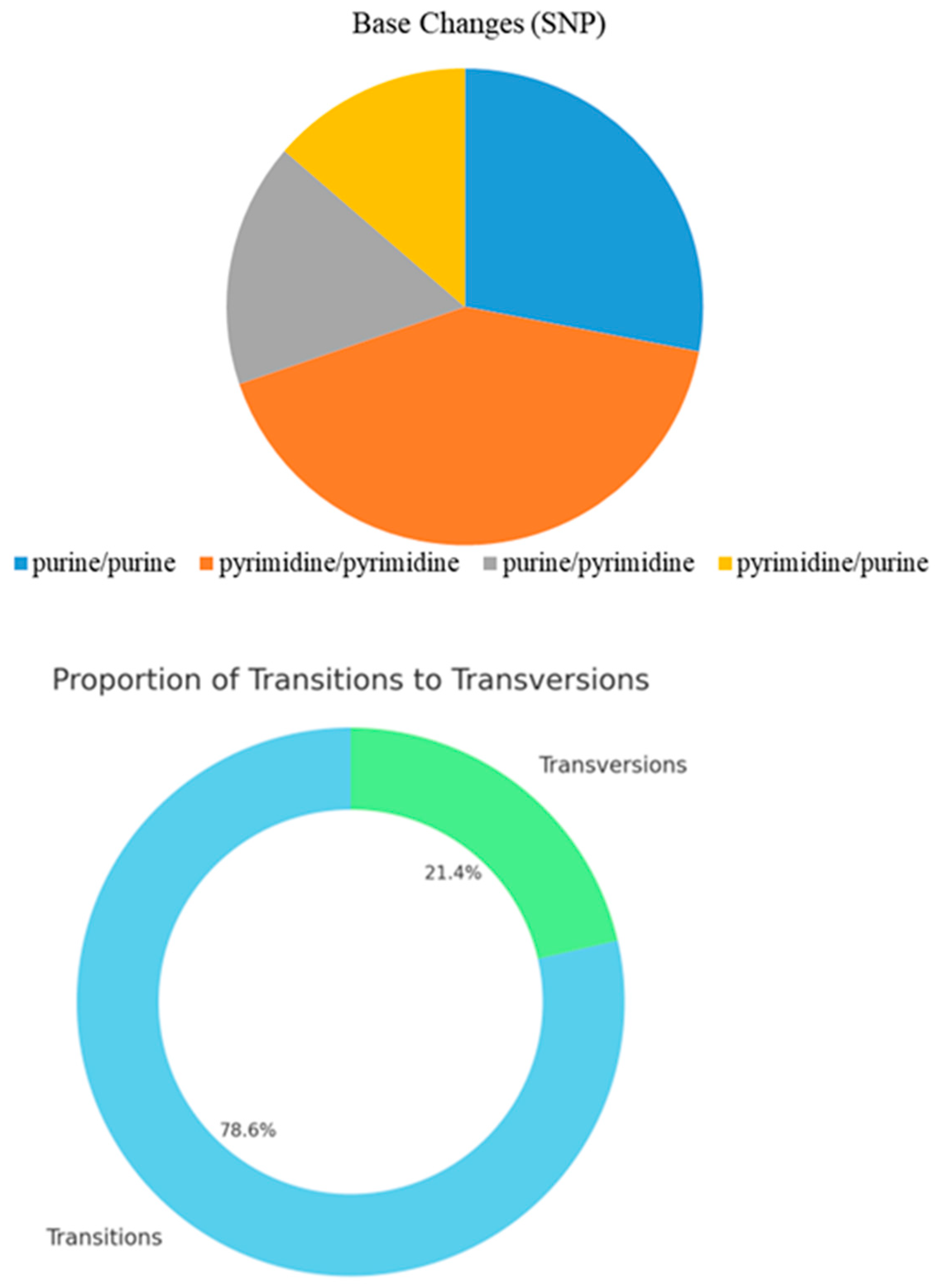
In this study, through WGR of 49 tropical oil tea camellia germplasm resources, 247,867,204 SNPs and 25,687,017 InDel variation sites were obtained with 96.60% alignment with the reference genome. After filtering out low-quality reads and excluding heterozygous and missing data, we obtained 239,441,603 high-quality SNPs and 23,510,374 high-quality InDels. A variation analysis of 49 oil tea camellia samples using these SNP and InDel loci, which cover the whole genome, revealed that there were significant differences in the distribution of SNPs and InDels in various functional regions, with a higher distribution in the intergenic and upstream regions. This suggests that there is greater evolutionary variability in these regions, and the SNP and InDel variations in the non-coding regions may have a significant impact on oil tea camellia gene expression regulation and genetic diversity. In contrast, variations in the genomic sequences of the tested samples were low in the coding regions. This may have occurred because the variation in coding regions tends to cause deleterious mutations, which are eliminated by strong selection during evolution. Based on our analysis of base mutations at SNP sites, we found that transitions were the predominant type of SNP variation in the genome of this sample. This finding aligns with the typical mutation pattern in most species, where transitions are more likely to occur and be retained than transversions due to complementary base pairing constraints.
The results of this study provide valuable insights into the genetic diversity and variation patterns of oil tea camellia, particularly in tropical populations. The high-quality SNP and InDel datasets generated here offer a valuable resource for future research, including the identification of key genetic markers for breeding programs aimed at improving ecological adaptability, oil yield, and resistance to environmental stresses. Furthermore, our findings highlight the importance of non-coding regions in maintaining genetic diversity, suggesting that future studies should focus on the functional impact of variations in these regions, especially their role in gene regulation and adaptation mechanisms. Advances in sequencing technologies and bioinformatics approaches will be essential for further reducing detection errors and enhancing the understanding of the complex genomic architecture of oil tea camellia. Such efforts will contribute significantly to the conservation and genetic improvement of this economically important species.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}