
Genome Assembly and Structural Variation Analysis of Luffa acutangula Provide Insights on Flowering Time and Ridge Development

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
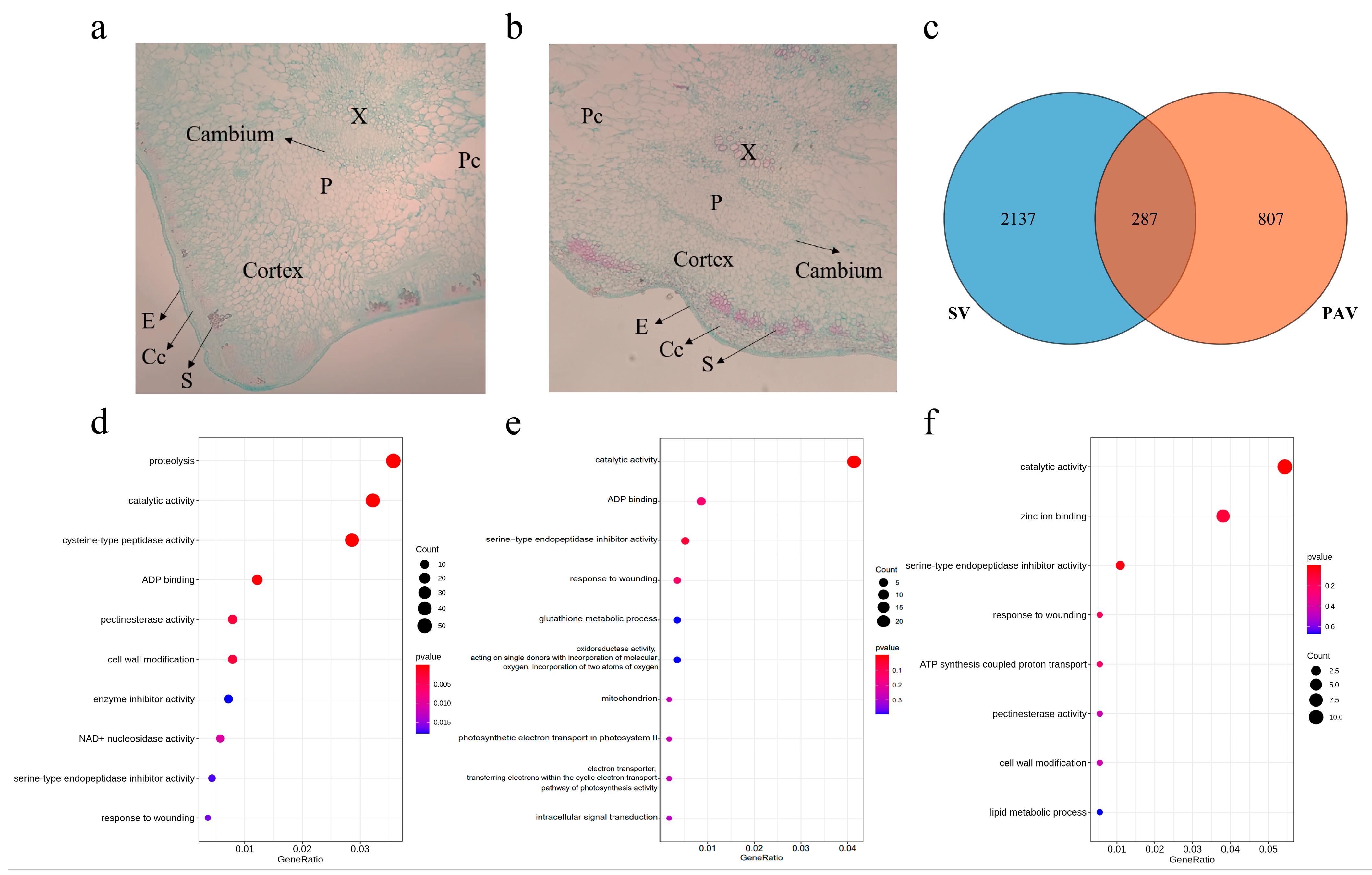
2.2. Section Observation
2.3. Genome Sequencing
2.4. RNA-Seq Library Construction and Sequencing
2.5. De Novo Sequencing and Genome Assembly
2.6. Repetitive Element Annotation and Gene Prediction
2.7. Comparative Genome Analysis
2.8. Structural Variants Analysis
2.9. GO Enrichment and Functional Annotation of Genes with SVs and PAVs
2.10. Analysis of Expansion and Contraction for Flowering-Time-Related Genes
3. Results and Discussion
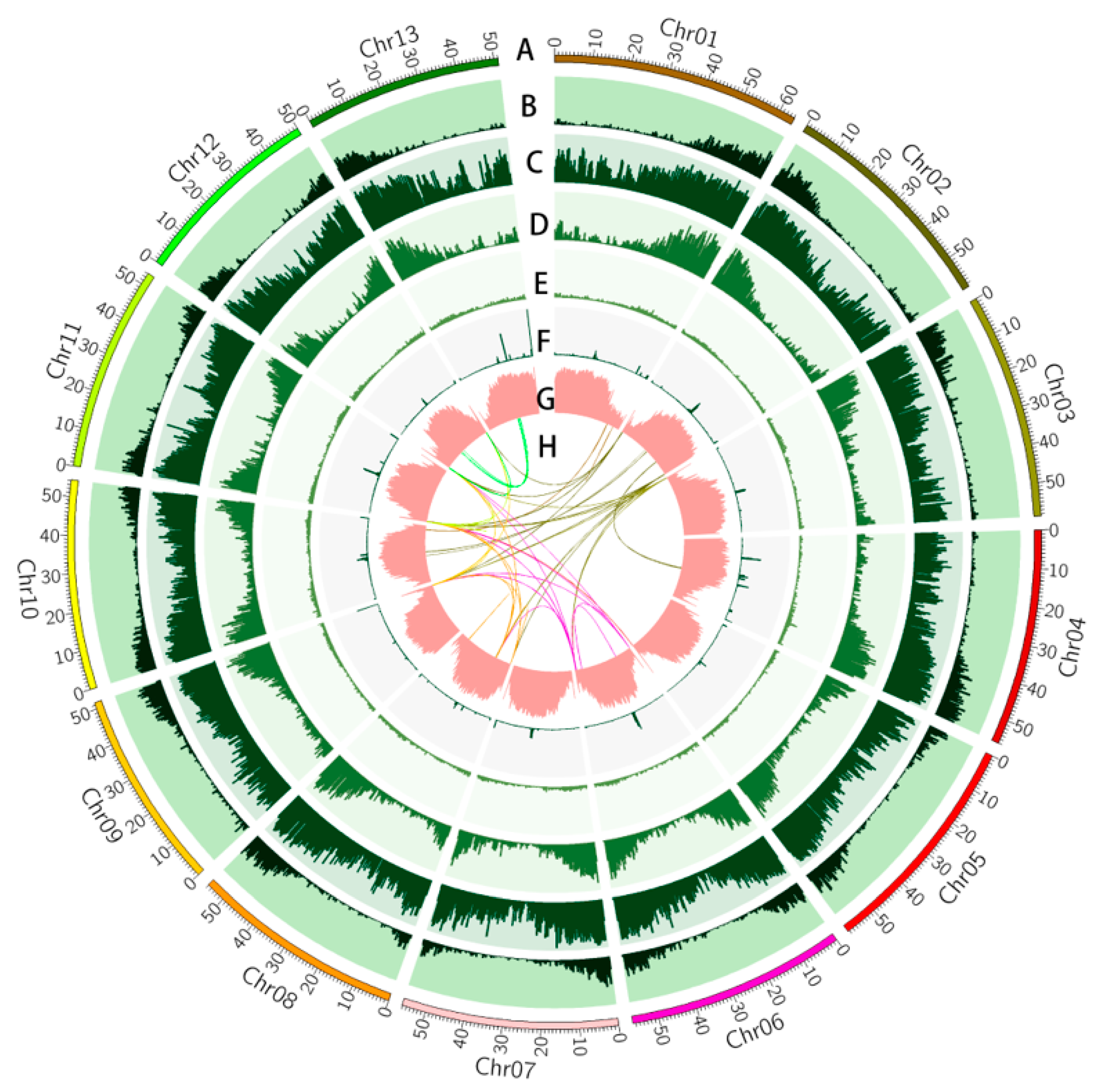
3.1. Genome Sequencing and Assembly
3.2. Genome Annotation
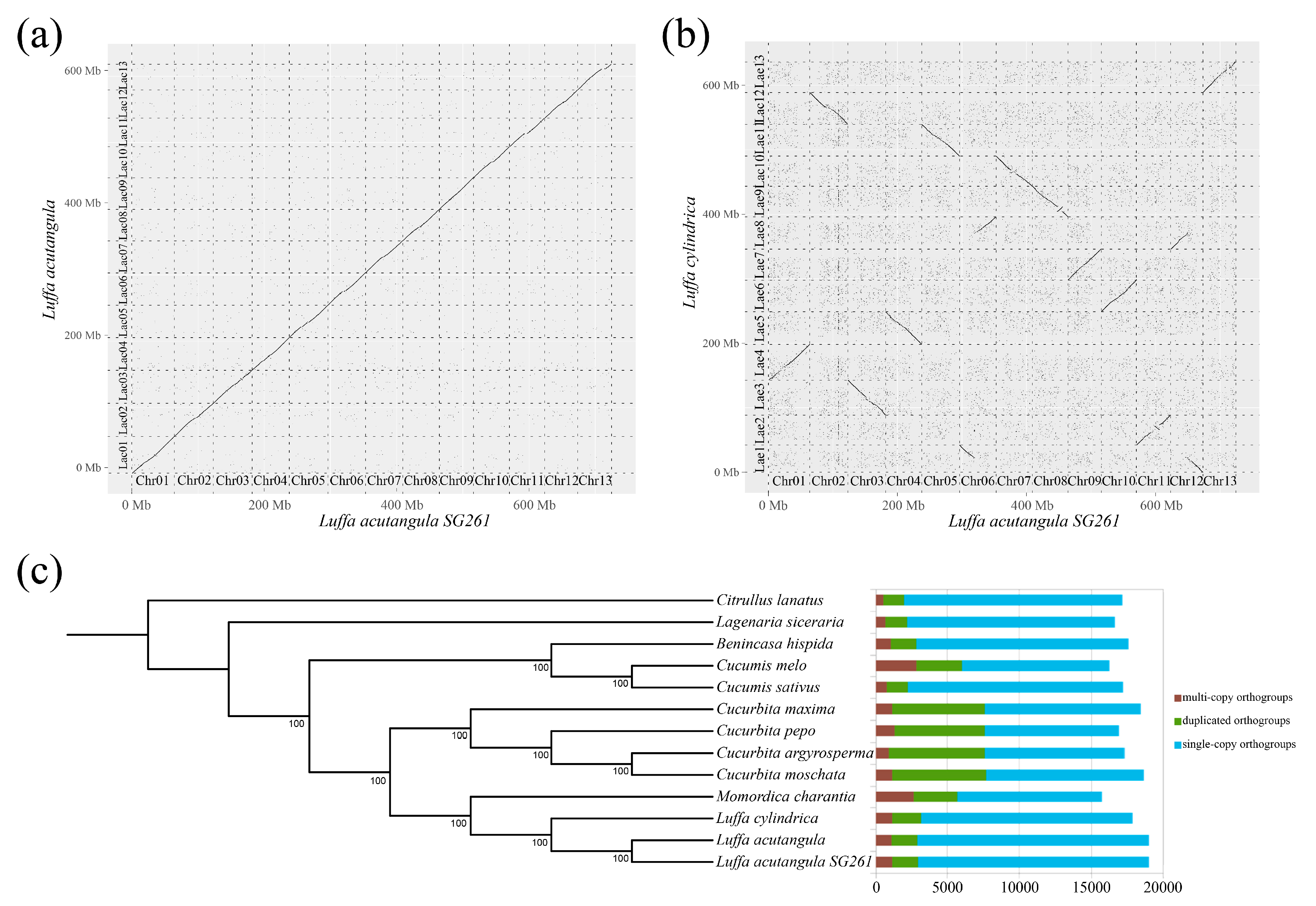
3.3. Synteny and Phylogenetic Analysis
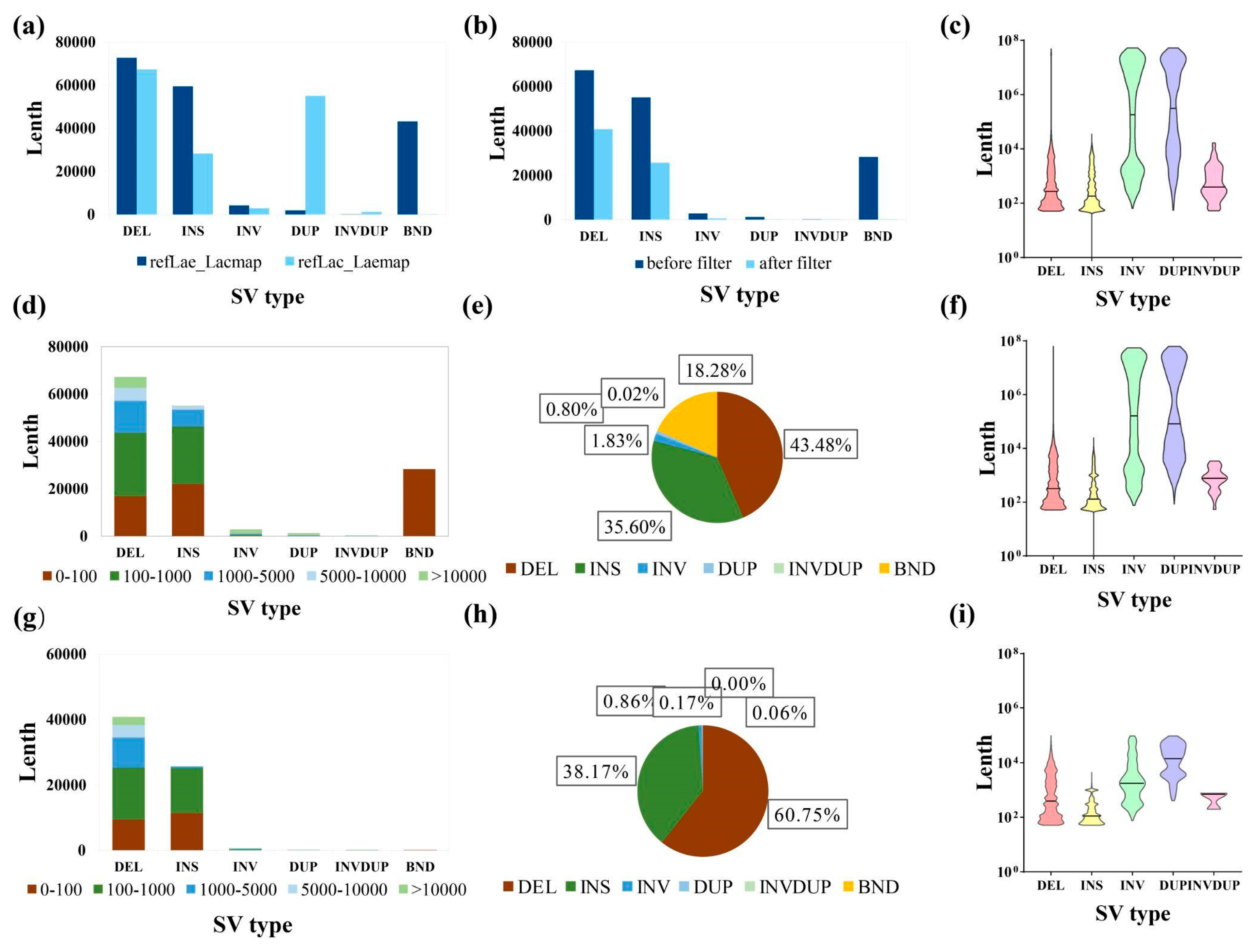
3.4. Variation between Luffa acutangula SG261 and Luffa cylindrica
3.5. Functional Annotation and GO Enrichment of SV and PAV Genes
3.6. Structural Variation Genes Involved in Ridge Development
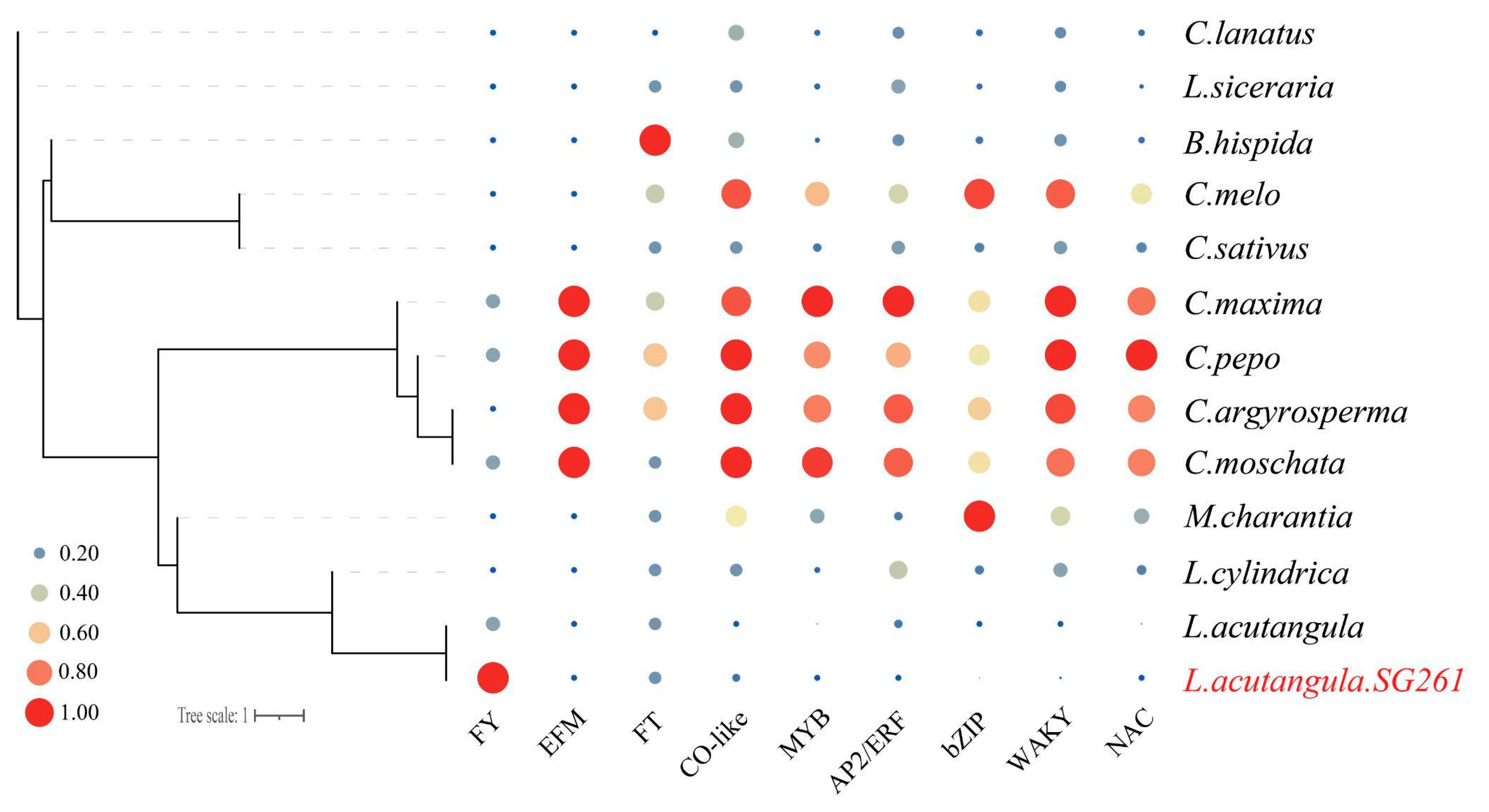
3.7. Analysis of Flowering-Time-Related Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Filipowicz, N.; Schaefer, H.; Renner, S.S. Revisiting Luffa (Cucurbitaceae) 25 Years after C. Heiser: Species Boundaries and Application of Names Tested with Plastid and Nuclear DNA Sequences. Syst. Bot. 2014, 39, 205–215. [Google Scholar] [CrossRef]
- Kalloo, G. Loofah. In Genetic Improvement of Vegetable Crops; Elsevier: Amsterdam, The Netherlands, 1993; pp. 265–266. ISBN 9780080408262. [Google Scholar]
- Prakash, K.; Pandey, A.; Radhamani, J.; Bisht, I.S. Morphological Variability in Cultivated and Wild Species of Luffa (Cucurbitaceae) from India. Genet. Resour. Crop Evol. 2013, 60, 2319–2329. [Google Scholar] [CrossRef]
- Islam, S.; Munshi, A.D.; Mandal, B.; Kumar, R.; Behera, T.K. Genetics of Resistance in Luffa cylindrica Roem. against Tomato Leaf Curl New Delhi Virus. Euphytica 2010, 174, 83–89. [Google Scholar] [CrossRef]
- Rabei, S.; Rizk, R.M.; Khedr, A.-H.A. Keys for and Morphological Character Variation in Some Egyptian Cultivars of Cucurbitaceae. Genet. Resour. Crop Evol. 2013, 60, 1353–1364. [Google Scholar] [CrossRef]
- Shendge, P.N.; Belemkar, S. Therapeutic Potential of Luffa acutangula: A Review on Its Traditional Uses, Phytochemistry, Pharmacology and Toxicological Aspects. Front. Pharmacol. 2018, 9, 1177. [Google Scholar]
- Kumari, S.A.S.M.; Nakandala, N.D.U.S.; Nawanjana, P.W.I.; Rathnayake, R.M.S.K.; Senavirathna, H.M.T.N.; Senevirathna, R.W.K.M.; Wijesundara, W.M.D.A.; Ranaweera, L.T.; Mannanayake, M.A.D.K.; Weebadde, C.K.; et al. The Establishment of the Species-Delimits and Varietal-Identities of the Cultivated Germplasm of Luffa acutangula and Luffa aegyptiaca in Sri Lanka Using Morphometric, Organoleptic and Phylogenetic Approaches. PLoS ONE 2019, 14, e0215176. [Google Scholar]
- Cui, J.; Cheng, J.; Wang, G.; Tang, X.; Wu, Z.; Lin, M.; Li, L.; Hu, K. QTL Analysis of Three Flower-Related Traits Based on an Interspecific Genetic Map of Luffa. Euphytica 2015, 202, 45–54. [Google Scholar]
- An, J.; Yin, M.; Zhang, Q.; Gong, D.; Jia, X.; Guan, Y.; Hu, J. Genome Survey Sequencing of Luffa cylindrica L. and Microsatellite High Resolution Melting (SSR-HRM) Analysis for Genetic Relationship of Luffa Genotypes. Int. J. Mol. Sci. 2017, 18, 1942. [Google Scholar] [CrossRef]
- Pootakham, W.; Sonthirod, C.; Naktang, C.; Nawae, W.; Yoocha, T.; Kongkachana, W.; Sangsrakru, D.; Jomchai, N.; U-Thoomporn, S.; Sheedy, J.R.; et al. De Novo Assemblies of Luffa acutangula and Luffa cylindrica Genomes Reveal an Expansion Associated with Substantial Accumulation of Transposable Elements. Mol. Ecol. Resour. 2021, 21, 212–225. [Google Scholar]
- Wu, H.; Zhao, G.; Gong, H.; Li, J.; Luo, C.; He, X.; Luo, S.; Zheng, X.; Liu, X.; Guo, J.; et al. A High-Quality Sponge Gourd (Luffa cylindrica) Genome. Hortic. Res. 2020, 7, 128. [Google Scholar] [CrossRef]
- Zhang, T.; Ren, X.; Zhang, Z.; Ming, Y.; Yang, Z.; Hu, J.; Li, S.; Wang, Y.; Sun, S.; Sun, K.; et al. Long-Read Sequencing and de Novo Assembly of the Luffa cylindrica (L.) Roem. Genome. Mol. Ecol. Resour. 2020, 20, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Alonge, M.; Wang, X.; Benoit, M.; Soyk, S.; Pereira, L.; Zhang, L.; Suresh, H.; Ramakrishnan, S.; Maumus, F.; Ciren, D.; et al. Major Impacts of Widespread Structural Variation on Gene Expression and Crop Improvement in Tomato. Cell 2020, 182, 145–161.e23. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.E.; Walter, K.; Stewart, C.; Handsaker, R.E.; Chen, K.; Alkan, C.; Abyzov, A.; Yoon, S.C.; Ye, K.; Cheetham, R.K.; et al. Mapping Copy Number Variation by Population-Scale Genome Sequencing. Nature 2011, 470, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Audano, P.A.; Sulovari, A.; Graves-Lindsay, T.A.; Cantsilieris, S.; Sorensen, M.; Welch, A.E.; Dougherty, M.L.; Nelson, B.J.; Shah, A.; Dutcher, S.K.; et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 2019, 176, 663–675.e19. [Google Scholar] [CrossRef]
- Li, H.; Wang, S.; Chai, S.; Yang, Z.; Zhang, Q.; Xin, H.; Xu, Y.; Lin, S.; Chen, X.; Yao, Z.; et al. Graph-Based Pan-Genome Reveals Structural and Sequence Variations Related to Agronomic Traits and Domestication in Cucumber. Nat. Commun. 2022, 13, 682. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Su, T.; Zhao, X.; Wang, W.; Zhang, D.; Yu, Y.; Bayer, P.E.; Edwards, D.; Yu, S.; Zhang, F. Assembly of the Non-heading Pak Choi Genome and Comparison with the Genomes of Heading Chinese Cabbage and the Oilseed Yellow Sarson. Plant Biotechnol. J. 2021, 19, 966–976. [Google Scholar] [CrossRef]
- Fischer, A.H.; Jacobson, K.A.; Rose, J.; Zeller, R. Paraffin embedding tissue samples for sectioning. CSH Protoc. 2008, 2008, prot4989. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.A.; Park, J.L.; Park, S.-J.; Kim, J.H.; Goh, S.-H.; Han, J.-Y.; Kim, S.-Y. Comparison between MGI and Illumina sequencing platforms for whole genome sequencing. Genes Genom. 2021, 43, 713–724. [Google Scholar] [CrossRef]
- Weirather, J.L.; de Cesare, M.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, X.J.; Au, K.F. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Research 2017, 6, 100. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce Acceleration-Supported Software for Integrated Quality Control and Preprocessing of High-Throughput Sequencing Data. Gigascience 2018, 7, gix120. [Google Scholar] [CrossRef]
- Liu, B.; Shi, Y.; Yuan, J.; Hu, X.; Zhang, H.; Li, N.; Li, Z.; Chen, Y.; Mu, D.; Fan, W. Estimation of Genomic Characteristics by Analyzing K-Mer Frequency in de Novo Genome Projects. arXiv 2013, arXiv:1308.2012. [Google Scholar] [CrossRef]
- Xiao, C.-L.; Chen, Y.; Xie, S.-Q.; Chen, K.-N.; Wang, Y.; Han, Y.; Luo, F.; Xie, Z. MECAT: Fast Mapping, Error Correction, and de Novo Assembly for Single-Molecule Sequencing Reads. Nat. Methods 2017, 14, 1072–1074. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and Accurate Long-Read Assembly via Adaptive -Mer Weighting and Repeat Separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.P.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P.; et al. De Novo Assembly of the Genome Using Hi-C Yields Chromosome-Length Scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Ramírez, F.; Bhardwaj, V.; Arrigoni, L.; Lam, K.C.; Grüning, B.A.; Villaveces, J.; Habermann, B.; Akhtar, A.; Manke, T. High-Resolution TADs Reveal DNA Sequences Underlying Genome Organization in Flies. Nat. Commun. 2018, 9, 189. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking Transposable Element Annotation Methods for Creation of a Streamlined, Comprehensive Pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef]
- Benson, G. Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Korf, I.; Robb, S.M.C.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Sánchez Alvarado, A.; Yandell, M. MAKER: An Easy-to-Use Annotation Pipeline Designed for Emerging Model Organism Genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro Protein Families and Domains Database: 20 Years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Xie, D.; Xu, Y.; Wang, J.; Liu, W.; Zhou, Q.; Luo, S.; Huang, W.; He, X.; Li, Q.; Peng, Q.; et al. The Wax Gourd Genomes Offer Insights into the Genetic Diversity and Ancestral Cucurbit Karyotype. Nat. Commun. 2019, 10, 5158. [Google Scholar] [CrossRef]
- Guo, S.; Zhao, S.; Sun, H.; Wang, X.; Wu, S.; Lin, T.; Ren, Y.; Gao, L.; Deng, Y.; Zhang, J.; et al. Resequencing of 414 Cultivated and Wild Watermelon Accessions Identifies Selection for Fruit Quality Traits. Nat. Genet. 2019, 51, 1616–1623. [Google Scholar] [CrossRef]
- Garcia-Mas, J.; Benjak, A.; Sanseverino, W.; Bourgeois, M.; Mir, G.; González, V.M.; Hénaff, E.; Câmara, F.; Cozzuto, L.; Lowy, E.; et al. The Genome of Melon (Cucumis melo L.). Proc. Natl. Acad. Sci. USA 2012, 109, 11872–11877. [Google Scholar] [CrossRef]
- Li, Q.; Li, H.; Huang, W.; Xu, Y.; Zhou, Q.; Wang, S.; Ruan, J.; Huang, S.; Zhang, Z. A Chromosome-Scale Genome Assembly of Cucumber (Cucumis sativus L.). Gigascience 2019, 8, giz072. [Google Scholar] [CrossRef]
- Barrera-Redondo, J.; Ibarra-Laclette, E.; Vázquez-Lobo, A.; Gutiérrez-Guerrero, Y.T.; Sánchez de la Vega, G.; Piñero, D.; Montes-Hernández, S.; Lira-Saade, R.; Eguiarte, L.E. The Genome of Cucurbita Argyrosperma (Silver-Seed Gourd) Reveals Faster Rates of Protein-Coding Gene and Long Noncoding RNA Turnover and Neofunctionalization within Cucurbita. Mol. Plant 2019, 12, 506–520. [Google Scholar] [CrossRef]
- Sun, H.; Wu, S.; Zhang, G.; Jiao, C.; Guo, S.; Ren, Y.; Zhang, J.; Zhang, H.; Gong, G.; Jia, Z.; et al. Karyotype Stability and Unbiased Fractionation in the Paleo-Allotetraploid Cucurbita Genomes. Mol. Plant 2017, 10, 1293–1306. [Google Scholar] [CrossRef]
- Montero-Pau, J.; Blanca, J.; Bombarely, A.; Ziarsolo, P.; Esteras, C.; Martí-Gómez, C.; Ferriol, M.; Gómez, P.; Jamilena, M.; Mueller, L.; et al. De Novo Assembly of the Zucchini Genome Reveals a Whole-Genome Duplication Associated with the Origin of the Cucurbita Genus. Plant Biotechnol. J. 2018, 16, 1161–1171. [Google Scholar] [CrossRef]
- Wu, S.; Shamimuzzaman, M.; Sun, H.; Salse, J.; Sui, X.; Wilder, A.; Wu, Z.; Levi, A.; Xu, Y.; Ling, K.-S.; et al. The Bottle Gourd Genome Provides Insights into Cucurbitaceae Evolution and Facilitates Mapping of a Papaya Ring-Spot Virus Resistance Locus. Plant J. 2017, 92, 963–975. [Google Scholar] [CrossRef]
- Urasaki, N.; Takagi, H.; Natsume, S.; Uemura, A.; Taniai, N.; Miyagi, N.; Fukushima, M.; Suzuki, S.; Tarora, K.; Tamaki, M.; et al. Draft Genome Sequence of Bitter Gourd (Momordica charantia), a Vegetable and Medicinal Plant in Tropical and Subtropical Regions. DNA Res. 2017, 24, 51–58. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Mirarab, S.; Reaz, R.; Bayzid, M.S.; Zimmermann, T.; Swenson, M.S.; Warnow, T. ASTRAL: Genome-Scale Coalescent-Based Species Tree Estimation. Bioinformatics 2014, 30, i541–i548. [Google Scholar] [CrossRef]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast Processing of NGS Alignment Formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Narasimhan, V.; Danecek, P.; Scally, A.; Xue, Y.; Tyler-Smith, C.; Durbin, R. BCFtools/RoH: A Hidden Markov Model Approach for Detecting Autozygosity from next-Generation Sequencing Data. Bioinformatics 2016, 32, 1749–1751. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate Detection of Complex Structural Variations Using Single-Molecule Sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef]
- Zhang, W.; Jia, B.; Wei, C. PaSS: A Sequencing Simulator for PacBio Sequencing. BMC Bioinform. 2019, 20, 352. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Yang, X.; Li, X.; Liao, W. Advances in the Genetic Regulating Pathways of Plant Flowering Time. Sheng Wu Duo Yang Xing 2021, 29, 825–842. [Google Scholar] [CrossRef]
- Sato, H.; Heang, D.; Sassa, H.; Koba, T. Identification and Characterization of FT/TFL1 Gene Family in Cucumber. Breed. Sci. 2009, 59, 3–11. [Google Scholar] [CrossRef]
- Tian, Z.; Qin, X.; Wang, H.; Li, J.; Chen, J. Genome-Wide Identification and Expression Analyses of CONSTANS-Like Family Genes in Cucumber (Cucumis sativus L.). J. Plant Growth Regul. 2022, 41, 1627–1641. [Google Scholar] [CrossRef]
- Cheng, C.; Li, Q.; Wang, X.; Li, Y.; Qian, C.; Li, J.; Lou, Q.; Jahn, M.; Chen, J. Identification and Expression Analysis of the Gene Family in Root Knot Nematode-Resistant and Susceptible Cucumbers. Front. Genet. 2020, 11, 550677. [Google Scholar] [CrossRef]
- Lee, S.-C.; Lee, W.-K.; Ali, A.; Kumar, M.; Yang, T.-J.; Song, K. Genome-Wide Identification and Classification of the AP2/EREBP Gene Family in the Cucurbitaceae Species. Plant Breed. Biotechnol. 2017, 5, 123–133. [Google Scholar] [CrossRef]
- Baloglu, M.C.; Eldem, V.; Hajyzadeh, M.; Unver, T. Genome-Wide Analysis of the bZIP Transcription Factors in Cucumber. PLoS ONE 2014, 9, e96014. [Google Scholar] [CrossRef]
- Liu, X.; Wang, T.; Bartholomew, E.; Black, K.; Dong, M.; Zhang, Y.; Yang, S.; Cai, Y.; Xue, S.; Weng, Y.; et al. Comprehensive Analysis of NAC Transcription Factors and Their Expression during Fruit Spine Development in Cucumber (Cucumis sativus L.). Hortic. Res. 2018, 5, 31. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Pflug, J.M.; Holmes, V.R.; Burrus, C.; Johnston, J.S.; Maddison, D.R. Measuring Genome Sizes Using Read-Depth, K-Mers, and Flow Cytometry: Methodological Comparisons in Beetles (Coleoptera). G3 Genes Genomes Genet. 2020, 10, 3047–3060. [Google Scholar] [CrossRef]
- Chomicki, G.; Schaefer, H.; Renner, S.S. Origin and Domestication of Cucurbitaceae Crops: Insights from Phylogenies, Genomics and Archaeology. New Phytol. 2020, 226, 1240–1255. [Google Scholar] [CrossRef]
- Feulner, P.G.D.; Chain, F.J.J.; Panchal, M.; Eizaguirre, C.; Kalbe, M.; Lenz, T.L.; Mundry, M.; Samonte, I.E.; Stoll, M.; Milinski, M.; et al. Genome-Wide Patterns of Standing Genetic Variation in a Marine Population of Three-Spined Sticklebacks. Mol. Ecol. 2013, 22, 635–649. [Google Scholar] [CrossRef]
- Zhao, Z.; Fu, Y.-X.; Hewett-Emmett, D.; Boerwinkle, E. Investigating Single Nucleotide Polymorphism (SNP) Density in the Human Genome and Its Implications for Molecular Evolution. Gene 2003, 312, 207–213. [Google Scholar] [CrossRef]
- Levesque-Tremblay, G.; Pelloux, J.; Braybrook, S.A.; Müller, K. Tuning of Pectin Methylesterification: Consequences for Cell Wall Biomechanics and Development. Planta 2015, 242, 791–811. [Google Scholar] [CrossRef]
- Kitamoto, N.; Ueno, S.; Takenaka, A.; Tsumura, Y.; Washitani, I.; Ohsawa, R. Effect of Flowering Phenology on Pollen Flow Distance and the Consequences for Spatial Genetic Structure within a Population of Primula sieboldii (Primulaceae). Am. J. Bot. 2006, 93, 226–233. [Google Scholar] [CrossRef]
- Pin, P.A.; Nilsson, O. The Multifaceted Roles of FLOWERING LOCUS T in Plant Development. Plant Cell Environ. 2012, 35, 1742–1755. [Google Scholar] [CrossRef]
- Simpson, G.G. The Autonomous Pathway: Epigenetic and Post-Transcriptional Gene Regulation in the Control of Arabidopsis Flowering Time. Curr. Opin. Plant Biol. 2004, 7, 570–574. [Google Scholar] [CrossRef]
- Srikanth, A.; Schmid, M. Regulation of Flowering Time: All Roads Lead to Rome. Cell. Mol. Life Sci. 2011, 68, 2013–2037. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Illumina + PacBio + Hi-C | |||
|---|---|---|---|---|
| Length | Number | |||
| Contig (bp) | Scaffold (bp) | Contig | Scaffold | |
| Total | 739,313,328 | 739,817,328 | 283 | 31 |
| MAX | 37,192,690 | 64,188,378 | - | - |
| mean | 2,612,414 | 23,865,075 | - | - |
| A | 236,049,024 | 236,049,024 | 31.93% | 31.91% |
| C | 133,750,610 | 133,750,610 | 18.09% | 18.08% |
| T | 235,878,785 | 235,878,785 | 31.91% | 31.88% |
| G | 133,634,909 | 133,634,909 | 18.08% | 18.06% |
| N | - | 504,000 | - | 0.07% |
| N50 | 18,375,156 | 56,077,004 | 15 (5.30%) | 7 (22.58%) |
| N60 | 13,378,742 | 55,168,345 | 20 (7.07%) | 8 (25.81%) |
| N70 | 10,763,832 | 54,241,644 | 26 (9.19%) | 9 (29.03%) |
| N80 | 8,026,258 | 51,658,337 | 34 (12.01%) | 11 (35.48%) |
| N90 | 4,503,403 | 51,095,587 | 46 (16.25%) | 12 (38.71%) |
| Luffa cylindrica (L.) [12] | Luffa cylindrica (L.) [11] | Luffa Cylindrica [10] | Luffa acutangula [10] | Luffa acutangula SG261 | |
|---|---|---|---|---|---|
| Sequence method | Pacbio | Pacbio | Pacbio | Pacbio | Pacbio |
| N50 (contig) (bp) | 4,815,853 | 8,800,239 | - | 110,403 | 18,375,156 |
| N50 (scaffold) (bp) | 48,664,788 | 48,760,765 | 578,616 | 47,609,564 | 56,077,004 |
| Genome size (Mb) | 669.7 | 656.2 | 689.8 | 735.6 | 739.8 |
| Longest scaffold (bp) | 62,749,569 | 55,641,800 | 7,054,290 | 56,032,585 | 64,188,378 |
| Repetitive sequence length (bp) | 416,310,000 | 419,095,893 | 391,650,000 | 456,690,000 | 536,834,300 |
| Non-repeated sequence length (bp) | 253,398,411 | 237,094,093 | 298,222,192 | 278,920,612 | 202,983,028 |
| BUSCO | 91.6% | 95.5% | 93.00% | 92.70% | 91.25% |
| Total number of genes | 31,661 | 27,154 | 43,828 | 32,233 | 27,312 |
| Average transcript (mRNA) length (bp) | - | 4184.44 | - | 1508.632356 | 1350.665312 |
| Average CDS length (bp) | 1246.02 | 1160.18 | - | 1067.034598 | 1047.6968 |
| Average exon length (bp) | 218.87 | 241.63 | 258,1 | 233.5 | 294.6388316 |
| Average gene length (bp) | 4387.94 | 4734.773861 | 2582 | 2866 | 3404.439707 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, A.; Feng, S.; Ye, Z.; Zhang, T.; Chen, S.; Chen, C.; Chen, S. Genome Assembly and Structural Variation Analysis of Luffa acutangula Provide Insights on Flowering Time and Ridge Development. Plants 2024, 13, 1828. https://doi.org/10.3390/plants13131828
Huang A, Feng S, Ye Z, Zhang T, Chen S, Chen C, Chen S. Genome Assembly and Structural Variation Analysis of Luffa acutangula Provide Insights on Flowering Time and Ridge Development. Plants. 2024; 13(13):1828. https://doi.org/10.3390/plants13131828
Chicago/Turabian StyleHuang, Aizheng, Shuo Feng, Zhuole Ye, Ting Zhang, Shenglong Chen, Changming Chen, and Shijun Chen. 2024. "Genome Assembly and Structural Variation Analysis of Luffa acutangula Provide Insights on Flowering Time and Ridge Development" Plants 13, no. 13: 1828. https://doi.org/10.3390/plants13131828
APA StyleHuang, A., Feng, S., Ye, Z., Zhang, T., Chen, S., Chen, C., & Chen, S. (2024). Genome Assembly and Structural Variation Analysis of Luffa acutangula Provide Insights on Flowering Time and Ridge Development. Plants, 13(13), 1828. https://doi.org/10.3390/plants13131828