

Transcriptome Analysis of Stigmas of Vicia faba L. Flowers


Abstract
1. Introduction
2. Results
2.1. Transcriptome Sequencing and De Novo Assembly
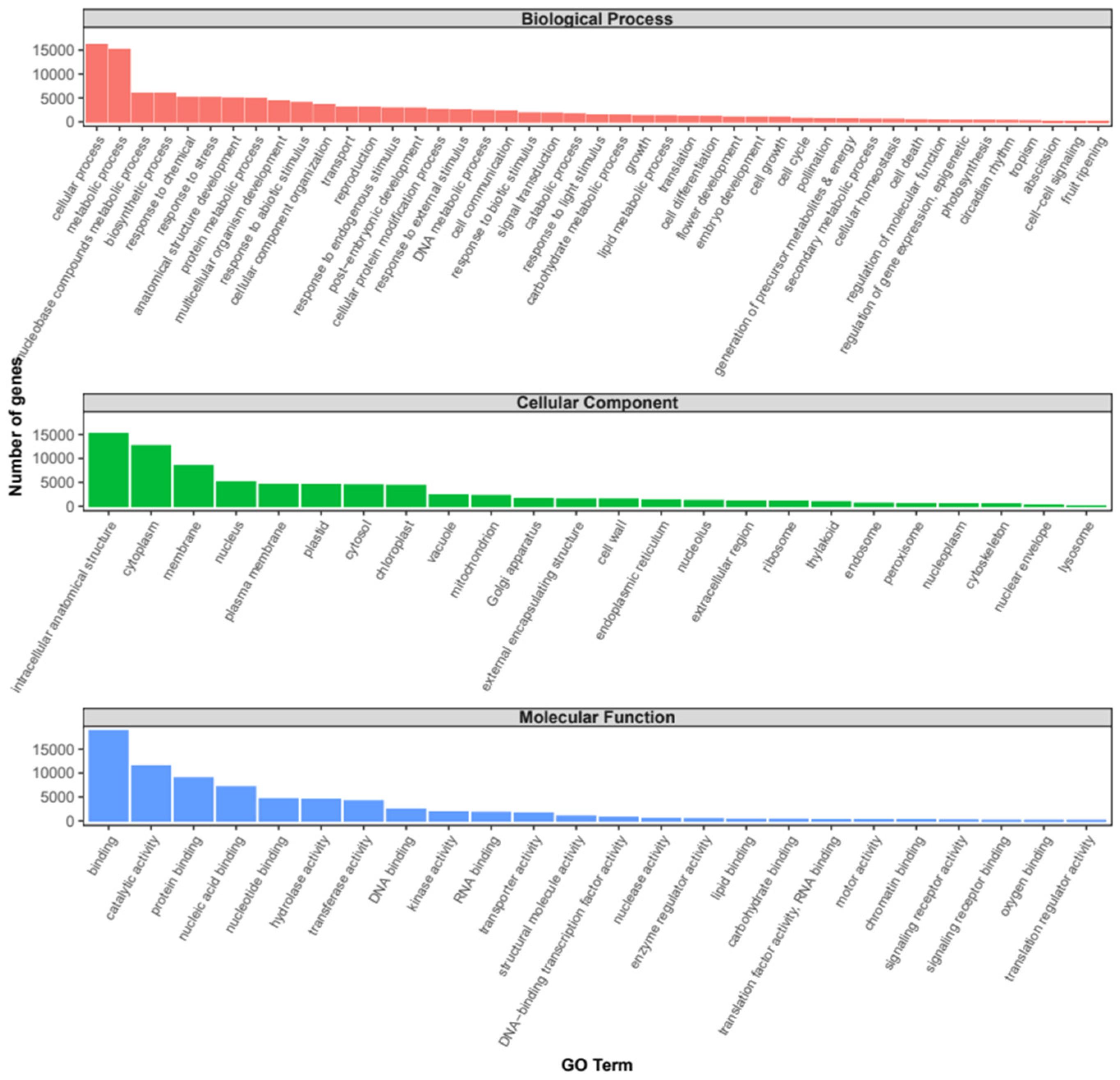
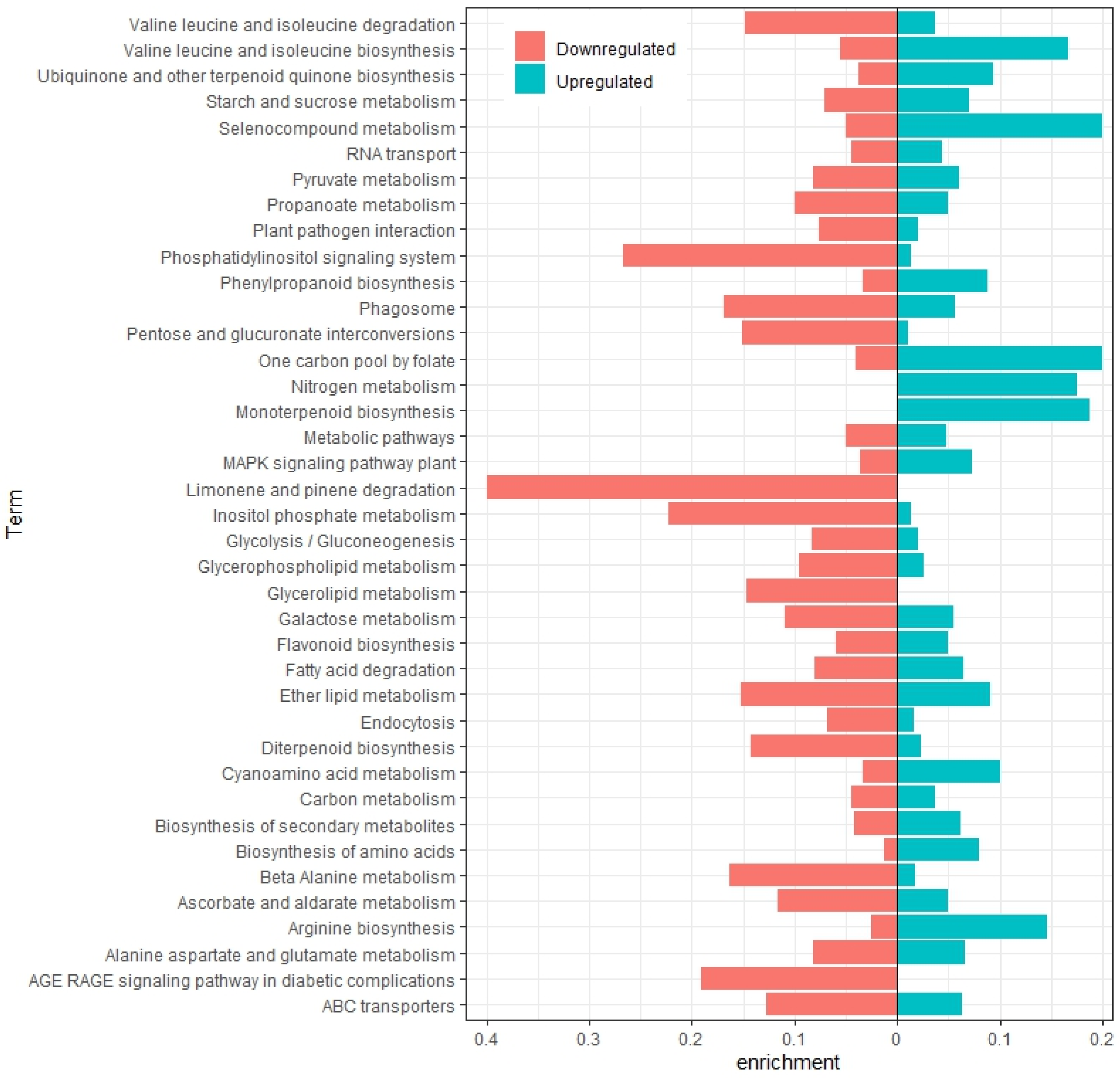
2.2. Annotation and Differential Expression Analysis
2.3. Search of DEGs within QTL Intervals for Autofertility
2.4. Quantitative Real-Time PCR Analysis
3. Discussion
3.1. Sequencing, Assembly, and Annotation
3.2. DEGs within QTL Intervals Previously Described for Autofertility
4. Materials and Methods
4.1. Plant Materials and Sample Collection
4.2. RNA Extraction, Sequencing, and De Novo Assembly
4.3. Annotation and Differential Expression Analysis
4.4. Search of DEGs within QTL Intervals Previously Described for Autofertility
4.5. Quantitative Real-Time PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heslop-Harrison, Y.; Shivanna, K.R. The receptive surface of the angiosperm stigma. Ann. Bot. 1977, 41, 1233–1258. [Google Scholar] [CrossRef]
- Heslop-Harrison, Y. Stigma characteristics and angiosperm taxonomy. Nord. J. Bot. 1981, 1, 401–420. [Google Scholar] [CrossRef]
- McInnis, S.M.; Costa, L.M.; Gutiérrez-Marcos, J.F.; Henderson, C.A.; Hiscock, S.J. Isolation and characterization of a polymorphic stigma-specific class III peroxidase gene from Senecio squalidus L. (Asteraceae). Plant Mol. Biol. 2005, 57, 659–677. [Google Scholar] [CrossRef] [PubMed]
- Hiscock, S.J.; Allen, A.M. Diverse cell signalling pathways regulate pollen-stigma interactions: The search for consensus. New Phytol. 2008, 179, 286–317. [Google Scholar] [CrossRef] [PubMed]
- Swanson, R.; Edlund, A.F.; Preuss, D. Species specificity in pollen-pistil interactions. Annu. Rev. Genet. 2004, 38, 793–818. [Google Scholar] [CrossRef] [PubMed]
- Zinkl, G.M.; Zwiebel, B.I.; Grier, D.G.; Preuss, D. Pollen-stigma adhesion in Arabidopsis: A species-specific interaction mediated by lipophilic molecules in the pollen exine. Development 1999, 126, 5431–5440. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.; Sollapura, V.; Couroux, P.; Sprott, D.; Ravensdale, M.; Routly, E.; Xing, T.; Robert, L.S. The Brassica mature pollen and stigma proteomes: Preparing to meet. Plant J. 2021, 107, 1546–1568. [Google Scholar] [CrossRef] [PubMed]
- Edlund, A.F.; Swanson, R.; Preuss, D. Pollen and stigma structure and function: The role of diversity in pollination. Plant Cell 2004, 16 (Suppl. S1), S84–S97. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.M.; Lexer, C.; Hiscock, S.J. Comparative analysis of pistil transcriptomes reveals conserved and novel genes expressed in dry, wet, and semidry stigmas. Plant Physiol. 2010, 154, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.L.; Xu, M.; Ma, F.F.; Chen, H.; Xu, X.H.; Gao, X.-Q.; Zhang, X.S. Comparative proteomic analysis reveals similar and distinct features of proteins in dry and wet stigmas. Proteomics 2012, 12, 1983–1998. [Google Scholar] [CrossRef]
- Kenicer, G. Legumes of the World. Edited by G. Lewis, B. Schrire, B. MacKinder & M. Lock. Royal Botanic Gardens, Kew. 2005. xiv + 577pp., colour photographs & line drawings. ISBN 1 900 34780 6. £55.00 (hardback). Edinb. J. Bot. 2006, 62, 195–196. [Google Scholar] [CrossRef]
- Chen, W.; Stoddard, F.L.; Baldwin, T.C. Developmental Regulation of Mannan, Arabinogalactan-Protein, and Pectic Epitopes in Pistils of Vicia faba (Faba Bean). Int. J. Plant Sci. 2006, 167, 919–932. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, L.; Nan, Z.; Wang, Y. Comparative transcriptional profiling provides insights into the evolution and development of the zygomorphic flower of Vicia sativa (Papilionoideae). PLoS ONE 2013, 8, e57338. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Datta, S.; Shaw, A.K.; Thimmaiah, M.R.; Satya, P.; Mitra, J.; Kar, G. De novo transcriptome of a bast fibre crop Crotalaria juncea reveals T2 ribonuclease genes to investigate late-acting self-rejection of pollens. Ind. Crops Prod. 2023, 202, 117064. [Google Scholar] [CrossRef]
- Heslop-Harrison, J.; Heslop-Harrison, Y. Pollen-Stigma Interaction in the Leguminosae: The Organization of the Stigma in Trifolium pratense L. Ann. Bot. 1983, 51, 571–583. [Google Scholar] [CrossRef]
- Lord, E.M.; Heslop-Harrison, Y. Pollen-Stigma Interaction in the Leguminosae: Stigma Organization and the Breeding System in Vicia faba L. Ann. Bot. 1984, 54, 827–836. [Google Scholar] [CrossRef]
- Costa, M.F.B.; Paulino, J.V.; Marinho, C.R.; Leite, V.G.; Pedersoli, G.D.; Teixeira, S.P. Stigma Diversity in Tropical Legumes with Considerations on Stigma Classification. Bot. Rev. 2014, 80, 1–29. [Google Scholar] [CrossRef]
- Azani, N.; Babineau, M.; Bailey, C.D.; Banks, H.; Barbosa Ariane, R.; Pinto, R.B.; Boatwright, J.S.; Borges, L.M.; Brown, G.K.; Bruneau, A.; et al. A new subfamily classification of the Leguminosae based on a taxonomically comprehensive phylogeny–The Legume Phylogeny Working Group (LPWG). Taxon 2017, 66, 44–77. [Google Scholar] [CrossRef]
- Graham, P.H.; Vance, C.P. Legumes: Importance and constraints to greater use. Plant Physiol. 2003, 131, 872–877. [Google Scholar] [CrossRef]
- Hedhly, A.; Hormaza, J.I.; Herrero, M. Global warming and sexual plant reproduction. Trends Plant Sci. 2009, 14, 30–36. [Google Scholar] [CrossRef]
- Stoddard, F.L. Climate change can affect crop pollination in unexpected ways. J. Exp. Bot. 2017, 68, 1819–1821. [Google Scholar] [CrossRef]
- Rasmont, P.; Franzen, M.; Lecocq, T.; Harpke, A.; Roberts, S.; Biesmeijer, J.C.; Castro, L.; Cederberg, B.; Dvorak, L.; Fitzpatrick, U.; et al. Climatic Risk and Distribution Atlas of European Bumblebees; Pensoft Publishers: Sofia, Bulgaria, 2015; Volume 10, pp. 1–236. [Google Scholar] [CrossRef]
- Memmott, J.; Craze, P.G.; Waser, N.M.; Price, M.V. Global warming and the disruption of plant-pollinator interactions. Ecol. Lett. 2007, 10, 710–717. [Google Scholar] [CrossRef]
- Settele, J.; Bishop, J.; Potts, S.G. Climate change impacts on pollination. Nat. Plants 2016, 2, 16092. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, F.L.; Bond, D.A. The pollination requirements of the faba bean. Bee World 1987, 68, 144–152. [Google Scholar] [CrossRef]
- Adcock, M.E.; Lawes, D.A. Self-fertility and the distribution of seed yield in Vicia faba L. Euphytica 1976, 25, 89–96. [Google Scholar] [CrossRef]
- Kambal, A.E.; Bond, D.A.; Toynbee-Clarke, G. A study on the pollination mechanism in field beans (Vicia faba L.). J. Agric. Sci. 1976, 87, 519. [Google Scholar] [CrossRef]
- Torres, A.M.; Moreno, M.T.; Cubero, J.I. Genetics of Six Components of Autofertility in Vicia faba. Plant Breed. 1993, 110, 220–228. [Google Scholar] [CrossRef]
- Drayner, J.M. Self- and cross-fertility in field beans (Vicia faba Linn.). J. Agric. Sci. 1959, 53, 387–403. [Google Scholar] [CrossRef]
- Chen, W. Pollination, Fertilization and Floral Traits Co-Segregating with Autofertility in Faba Bean. J. New Seeds 2009, 10, 14–30. [Google Scholar] [CrossRef]
- Aguilar-Benitez, D.; Casimiro-Soriguer, I.; Ferrándiz, C.; Torres, A.M. Study and QTL mapping of reproductive and morphological traits implicated in the autofertility of faba bean. BMC Plant Biol. 2022, 22, 175. [Google Scholar] [CrossRef]
- Aguilar-Benitez, D.; Gutierrez, N.; Casimiro-Soriguer, I.; Torres, A.M. High-density linkage map and fine QTL mapping of architecture, phenology and yield related traits in faba bean (Vicia faba L.). 2024; manuscript in preparation. [Google Scholar]
- Khan, M.A.; Alghamdi, S.S.; Ammar, M.H.; Sun, Q.; Teng, F.; Migdadi, H.M.; Al-Faifi, S.A. Transcriptome profiling of faba bean (Vicia faba L.) drought-tolerant variety hassawi-2 under drought stress using RNA sequencing. Electron. J. Biotechnol. 2019, 39, 15–29. [Google Scholar] [CrossRef]
- Lyu, J.I.; Ramekar, R.; Kim, J.M.; Hung, N.N.; Seo, J.S.; Kim, J.-B.; Choi, I.Y.; Park, K.C.; Kwon, S.J. Unraveling the complexity of faba bean (Vicia faba L.) transcriptome to reveal cold-stress-responsive genes using long-read isoform sequencing technology. Sci. Rep. 2021, 11, 21094. [Google Scholar] [CrossRef] [PubMed]
- Ocaña, S.; Seoane, P.; Bautista, R.; Palomino, C.; Claros, G.M.; Torres, A.M.; Madrid, E. Large-Scale Transcriptome Analysis in Faba Bean (Vicia faba L.) under Ascochyta fabae Infection. PLoS ONE 2015, 10, e0135143. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Perdomo, E.; Vidal, A.; Kreplak, J.; Duborjal, H.; Leveugle, M.; Duarte, J.; Desmetz, C.; Deulvot, C.; Raffiot, B.; Marget, P.; et al. Development of new genetic resources for faba bean (Vicia faba L.) breeding through the discovery of gene-based SNP markers and the construction of a high-density consensus map. Sci. Rep. 2020, 10, 6790. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chen, H.; Liu, C.; Li, L.; Liu, L.; Han, X.; Wan, Z.; Sha, A. Transcriptome profile analysis of two Vicia faba cultivars with contrasting salinity tolerance during seed germination. Sci. Rep. 2020, 10, 7250. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bian, X.-C.; Yang, F.; Chen, M.-X.; Das, D.; Zhu, X.-R.; Jiang, Y.; Zhang, J.; Cao, Y.Y.; Wu, C.F. Comprehensive transcriptome analysis of faba bean in response to vernalization. Planta 2019, 251, 22. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, R.; Liu, Y.; Hou, W.; Wang, X.; Miao, Y.; He, Y.; Ma, Y.; Li, G.; Wang, D.; et al. Development and application of the Faba_bean_130K targeted next-generation sequencing SNP genotyping platform based on transcriptome sequencing. Theor. Appl. Genet. 2021, 134, 3195–3207. [Google Scholar] [CrossRef]
- Hou, W.; Zhang, X.; Liu, Y.; Liu, Y.; Feng, B.L. RNA-Seq and genetic diversity analysis of faba bean (Vicia faba L.) varieties in China. PeerJ 2023, 11, e14259. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qi, X.; Chen, S.; Feng, J.; Chen, H.; Qin, Z.; Deng, Y. An integrated transcriptomic and proteomic approach to dynamically study the mechanism of pollen-pistil interactions during jasmine crossing. J. Proteom. 2021, 249, 104380. [Google Scholar] [CrossRef]
- He, Y.; Song, Q.; Chen, S.; Wu, Y.; Zheng, G.; Feng, J.; Yang, Z.; Lin, W.; Li, Y.; Chen, H. Transcriptome analysis of self- and cross-pollinated pistils revealing candidate unigenes of self-incompatibility in Camellia oleifera. J. Hortic. Sci. Biotechnol. 2020, 95, 19–31. [Google Scholar] [CrossRef]
- Quiapim, A.C.; Brito, M.S.; Bernardes, L.A.S.; Dasilva, I.; Malavazi, I.; DePaoli, H.C.; Molfetta-Machado, J.B.; Giuliatti, S.; Goldman, G.H.; Goldman, M.H.S. Analysis of the Nicotiana tabacum stigma/style transcriptome reveals gene expression differences between wet and dry stigma species. Plant Physiol. 2009, 149, 1211–1230. [Google Scholar] [CrossRef] [PubMed]
- Gorelova, V.; Ambach, L.; Rébeillé, F.; Stove, C.; Van Der Straeten, D. Folates in plants: Research advances and progress in crop biofortification. Front. Chem. 2017, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Dötterl, S.; Gershenzon, J. Chemistry, biosynthesis and biology of floral volatiles: Roles in pollination and other functions. Nat. Prod. Rep. 2023, 40, 1901–1937. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Song, Q. Responses of the pollinating wasp Ceratosolen solmsi marchali to odor variation between two floral stages of Ficus hispida. J. Chem. Ecol. 2008, 34, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Sanchez-Moreiras, A.M.; Abel, C.; Sohrabi, R.; Lee, S.; Gershenzon, J.; Tholl, D. The major volatile organic compound emitted from Arabidopsis thaliana flowers, the sesquiterpene (E)-β-caryophyllene, is a defense against a bacterial pathogen. New Phytol. 2012, 193, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Bruce, T.J.A.; Martin, J.L.; Smart, L.E.; Pickett, J.A. Development of semiochemical attractants for monitoring bean seed beetle, Bruchus rufimanus. Pest. Manag. Sci. 2011, 67, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef]
- Jamshed, M.; Sankaranarayanan, S.; Abhinandan, K.; Samuel, M.A. Stigma receptivity is controlled by functionally redundant MAPK pathway components in arabidopsis. Mol. Plant 2020, 13, 1582–1593. [Google Scholar] [CrossRef] [PubMed]
- McInnis, S.M.; Desikan, R.; Hancock, J.T.; Hiscock, S.J. Production of reactive oxygen species and reactive nitrogen species by angiosperm stigmas and pollen: Potential signalling crosstalk? New Phytol. 2006, 172, 221–228. [Google Scholar] [CrossRef]
- Xie, J.; Méndez, J.D.; Méndez-Valenzuela, V.; Aguilar-Hernández, M.M. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef]
- Shumilina, J.; Kusnetsova, A.; Tsarev, A.; Janse van Rensburg, H.C.; Medvedev, S.; Demidchik, V.; Van den Ende, W.; Frolov, A. Glycation of Plant Proteins: Regulatory Roles and Interplay with Sugar Signalling? Int. J. Mol. Sci. 2019, 20, 2366. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.-W.; Chen, X.; Mei, Y. Function and regulation of phospholipid signalling in plants. Biochem. J. 2009, 421, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Thole, J.M.; Nielsen, E. Phosphoinositides in plants: Novel functions in membrane trafficking. Curr. Opin. Plant Biol. 2008, 11, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Preuss, M.L.; Schmitz, A.J.; Thole, J.M.; Bonner, H.K.S.; Otegui, M.S.; Nielsen, E. A role for the RabA4b effector protein PI-4Kbeta1 in polarized expansion of root hair cells in Arabidopsis thaliana. J. Cell Biol. 2006, 172, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yan, A.; Feijó, J.A.; Furutani, M.; Takenawa, T.; Hwang, I.; Fu, Y.; Yang, Z. Phosphoinositides regulate clathrin-dependent endocytosis at the tip of pollen tubes in Arabidopsis and tobacco. Plant Cell. 2010, 22, 4031–4044. [Google Scholar] [CrossRef] [PubMed]
- Noack, L.C.; Jaillais, Y. Functions of anionic lipids in plants. Annu. Rev. Plant Biol. 2020, 71, 71–102. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Watari, M.; Tsuge, T.; Zhong, S.; Gu, H.; Qu, L.-J.; Fujiwara, T.; Aoyama, T. Redundant function of the Arabidopsis phosphatidylinositol 4-phosphate 5-kinase genes PIP5K4-6 is essential for pollen germination. Plant J. 2024, 117, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Menzel, W.; Stenzel, I.; Helbig, L.-M.; Krishnamoorthy, P.; Neumann, S.; Eschen-Lippold, L.; Heilmann, M.; Lee, J.; Heilmann, I. A PAMP-triggered MAPK cascade inhibits phosphatidylinositol 4,5-bisphosphate production by PIP5K6 in Arabidopsis thaliana. New Phytol. 2019, 224, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Funck, D.; Eckard, S.; Müller, G. Non-redundant functions of two proline dehydrogenase isoforms in Arabidopsis. BMC Plant Biol. 2010, 10, 70. [Google Scholar] [CrossRef]
- Phang, J.M.; Pandhare, J.; Liu, Y. The metabolism of proline as microenvironmental stress substrate. J. Nutr. 2008, 138, 2008S–2015S. [Google Scholar] [CrossRef]
- Nagano, T.; Nakashima, A.; Onishi, K.; Kawai, K.; Awai, Y.; Kinugasa, M.; Iwasaki, T.; Kikkawa, U.; Kamada, S. Proline dehydrogenase promotes senescence through the generation of reactive oxygen species. J. Cell Sci. 2017, 130, 1413–1420. [Google Scholar] [CrossRef] [PubMed]
- Bauduin, S.; Latini, M.; Belleggia, I.; Migliore, M.; Biancucci, M.; Mattioli, R.; Francioso, A.; Mosca, L.; Funck, D.; Trovato, M. Interplay between Proline Metabolism and ROS in the Fine Tuning of Root-Meristem Size in Arabidopsis. Plants 2022, 11, 1512. [Google Scholar] [CrossRef] [PubMed]
- Bultreys, A.; Trombik, T.; Drozak, A.; Boutry, M. Nicotiana plumbaginifolia plants silenced for the ATP-binding cassette transporter gene NpPDR1 show increased susceptibility to a group of fungal and oomycete pathogens. Mol. Plant Pathol. 2009, 10, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Blaylock, L.A.; Harrison, M.J. Two Medicago truncatula half-ABC transporters are essential for arbuscule development in arbuscular mycorrhizal symbiosis. Plant Cell 2010, 22, 1483–1497. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Li, H.; Xia, X.; Meng, Y.; Wang, N.; Li, L.; Shi, J.; Pei, Y.; Lin, M.; Niu, L.; et al. ATP-Binding Cassette G Transporters SGE1 and MtABCG13 Control Stigma Exsertion. Plant Physiol. 2020, 184, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Do, T.H.T.; Choi, H.; Palmgren, M.; Martinoia, E.; Hwang, J.-U.; Lee, Y. Arabidopsis ABCG28 is required for the apical accumulation of reactive oxygen species in growing pollen tubes. Proc. Natl. Acad. Sci. USA 2019, 116, 12540–12549. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Benitez, D.; Casimiro-Soriguer, I.; Torres, A.M. First approach to pod dehiscence in faba bean: Genetic and histological analyses. Sci. Rep. 2020, 10, 17678. [Google Scholar] [CrossRef]
- Aguilar-Benitez, D.; Casimiro-Soriguer, I.; Maalouf, F.; Torres, A.M. Linkage mapping and QTL analysis of flowering time in faba bean. Sci. Rep. 2021, 11, 13716. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Bucchini, F.; Del Cortona, A.; Kreft, Ł.; Botzki, A.; Van Bel, M.; Vandepoele, K. TRAPID 2.0, a web application for taxonomic and functional analysis of de novo transcriptomes. Nucleic Acids Res. 2021, 49, e101. [Google Scholar] [CrossRef]
- Van Bel, M.; Diels, T.; Vancaester, E.; Kreft, L.; Botzki, A.; Van de Peer, Y.; Coppens, F.; Vandepoele, K. PLAZA 4.0, an integrative resource for functional, evolutionary and comparative plant genomics. Nucleic Acids Res. 2018, 46, D1190–D1196. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, D.M.; Angra, D.; Harvie, T.; Tagkouli, V.; Warsame, A. A genetic toolbox for Vicia faba improvement. In Proceedings of the International Conference on Legume Genetics and Genomics, Dijon, France, 13–17 May 2019. [Google Scholar]
- Khazaei, H.; O’Sullivan, D.M.; Stoddard, F.L.; Adhikari, K.N.; Paull, J.G.; Schulman, A.H.; Andersen, S.U.; Vandenberg, A. Recent advances in faba bean genetic and genomic tools for crop improvement. Legume Sci. 2021, 3, e75. [Google Scholar] [CrossRef] [PubMed]
- Jayakodi, M.; Golicz, A.A.; Kreplak, J.; Fechete, L.I.; Angra, D.; Bednář, P.; Bornhofen, E.; Zhang, H.; Boussageon, R.; Kaur, S.; et al. The giant diploid faba genome unlocks variation in a global protein crop. Nature 2023, 615, 652–659. [Google Scholar] [CrossRef]
- Gutierrez, N.; Giménez, M.J.; Palomino, C.; Avila, C.M. Assessment of candidate reference genes for expression studies in Vicia faba L. by real-time quantitative PCR. Mol. Breed. 2011, 28, 13–24. [Google Scholar] [CrossRef]
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Sample | Raw Reads | Number of Bases (Gb) | Clean Reads |
|---|---|---|---|
| AF27.18 | 133,319,602 | 19,997 | 121,359,412 |
| AF27.28 | 65,839,438 | 9875 | 60,315,740 |
| AF27.5 | 61,476,838 | 9221 | 56,683,402 |
| AF44.1 | 55,001,874 | 8250 | 49,763,678 |
| AF44.4 | 65,871,434 | 9880 | 59,782,290 |
| AF44.14 | 64,747,566 | 9712 | 59,231,922 |
| AF14.2 | 56,963,200 | 8544 | 43,002,584 |
| AF14.5 | 70,837,140 | 10,625 | 65,081,586 |
| AF14.16 | 63,834,224 | 9575 | 58,881,026 |
| AS6.3 | 73,167,636 | 10,975 | 67,520,844 |
| AS6.5 | 57,074,180 | 8561 | 52,288,334 |
| AS6.14 | 47,358,170 | 7103 | 42,875,694 |
| AS96.1 | 68,186,700 | 10,228 | 62,004,170 |
| AS96.9 | 63,583,396 | 9537 | 58,218,214 |
| AS96.12 | 57,687,624 | 8653 | 51,643,386 |
| AS19.2 | 57,687,624 | 9500 | 58,475,436 |
| AS19.3 | 63,147,560 | 9472 | 57,224,622 |
| AS19.9 | 57,643,612 | 8646 | 52,847,570 |
| Total | 1,189,079,630 | 1,077,199,910 |
| Transcript ID | Regulation | Traits | Chr | QTL Marker | Reference Genome Vicia Faba | Organism | Protein ID | Protein Description | Arabidopsis ID |
|---|---|---|---|---|---|---|---|---|---|
| TRINITY_DN9113_c0_g1 | UR | PSF_2009/10 | CHR1 | AX_416823680 | 1g073840 | Pisum sativum | XP_050883094.1 | protochlorophyllide reductase, chloroplastic | AT5G54190 |
| TRINITY_DN9113_c0_g1 | UR | PSF_2009/10 | CHR1 | AX_181440809 | 1g073840 | Pisum sativum | XP_050883094.1 | protochlorophyllide reductase, chloroplastic | AT5G54190 |
| TRINITY_DN239_c0_g2 | UR | RUPTL | CHR1 | AX_181440664 | 1g128360 | Pisum sativum | XP_050907114.1 | proline dehydrogenase 2, mitochondrial-like | AT5G38710 |
| TRINITY_DN1719_c0_g2 | UR | PSC_2008/09 | CHR4 | AX_416818743 | 4g037560 | Pisum sativum | XP_050882017.1 | DNA damage-repair/toleration protein DRT100-like | AT3G12610 |
| TRINITY_DN27765_c0_g4 | DR | PSC_2008/09 | CHR4 | AX_416752208 | 4g039440 | Pisum sativum | XP_050873114.1 | phosphatidylinositol 4-phosphate 5-kinase 6-like | AT3G07960 |
| TRINITY_DN10484_c0_g3 | DR | PSC_2008/09 | CHR4 | AX_416793113 | 4g042680 | Pisum sativum | XP_050873201.1 | probable receptor-like protein kinase At4g39110 | AT4G39110 |
| TRINITY_DN16420_c1_g1 | DR | PSC_2008/09 | CHR4 | AX_181170326 | 4g042600 | Pisum sativum | XP_050873193.1 | protein CHROMATIN REMODELING 35 | AT2G16390 |
| TRINITY_DN21326_c1_g1 | DR | PAPL | CHR4 | AX_416773786 | 4g230040 | Pisum sativum | XP_050920775.1 | N6-mAMP deaminase-like | AT4G04880 |
| TRINITY_DN1073_c0_g1 | UR | NPAP/STIGL | CHR6 | AX_416766831 | 6g033880 | Trifolium repens | KAK2436160.1 | DNA-binding protein | AT5G59830 |
| TRINITY_DN42106_c0_g1 | DR | %RUPTAREA | CHR6 | AX_416739703 | 6g026920 | Pisum sativum | XP_050899031.1 | ABC transporter G family member 28 | AT5G60740 |
| TRINITY_DN47868_c0_g1 | DR | %RUPTAREA | CHR6 | AX_416739703 | 6g026920 | Pisum sativum | XP_050899031.1 | ABC transporter G family member 28 | AT5G60740 |
| TRINITY_DN52070_c0_g1 | DR | %RUPTAREA | CHR6 | AX_416739703 | 6g026920 | Pisum sativum | XP_050899031.1 | ABC transporter G family member 28 | AT5G60740 |
| TRINITY_DN61450_c0_g1 | UR | SSC_2008/09 + OL | CHR6 | AX_181176378 | 6g125280 | Trifolium repens | KAK2434246.1 | plastid movement impaired protein | AT2G01340 |
| TRINITY_DN1763_c0_g2 | DR | SSC_2008/09 + OL | CHR6 | AX_181445379 | 6g128440 | Pisum sativum | XP_050893915.1 | xyloglucan endotransglucosylase/hydrolase 2 | AT3G23730 |
| TRINITY_DN1344_c0_g4 | DR | SSC_2008/09 + OL | CHR6 | AX_181445379 | 6g128440 | Pisum sativum | XP_050893915.1 | xyloglucan endotransglucosylase/hydrolase 2 | AT3G23730 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casimiro-Soriguer, I.; Aguilar-Benitez, D.; Gutierrez, N.; Torres, A.M. Transcriptome Analysis of Stigmas of Vicia faba L. Flowers. Plants 2024, 13, 1443. https://doi.org/10.3390/plants13111443
Casimiro-Soriguer I, Aguilar-Benitez D, Gutierrez N, Torres AM. Transcriptome Analysis of Stigmas of Vicia faba L. Flowers. Plants. 2024; 13(11):1443. https://doi.org/10.3390/plants13111443
Chicago/Turabian StyleCasimiro-Soriguer, Inés, David Aguilar-Benitez, Natalia Gutierrez, and Ana M. Torres. 2024. "Transcriptome Analysis of Stigmas of Vicia faba L. Flowers" Plants 13, no. 11: 1443. https://doi.org/10.3390/plants13111443
APA StyleCasimiro-Soriguer, I., Aguilar-Benitez, D., Gutierrez, N., & Torres, A. M. (2024). Transcriptome Analysis of Stigmas of Vicia faba L. Flowers. Plants, 13(11), 1443. https://doi.org/10.3390/plants13111443