
Comparative Genome-Wide Identification of the Fatty Acid Desaturase Gene Family in Tea and Oil Tea

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
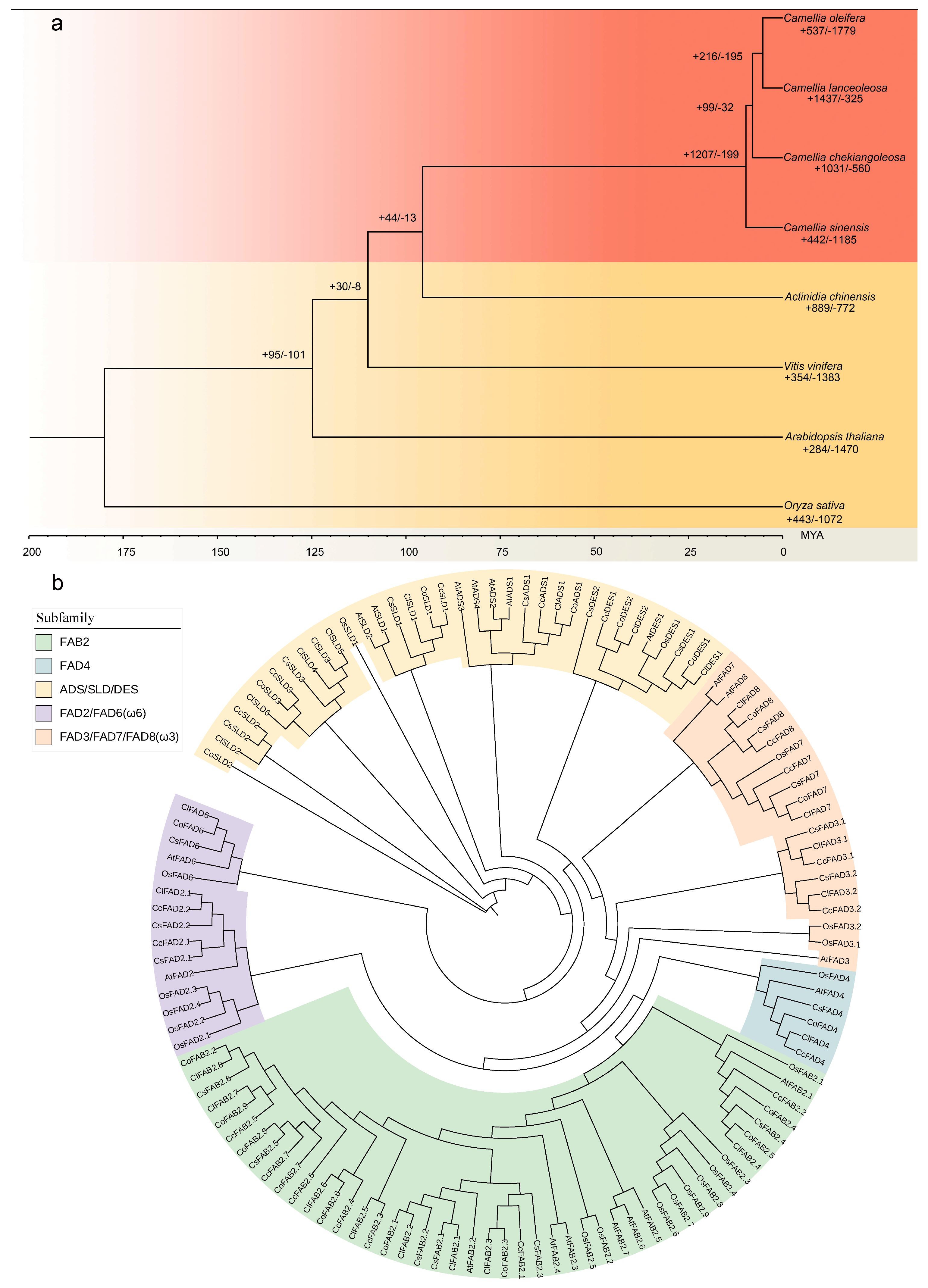
2.1. Genome Evolution and Expansion Analyses
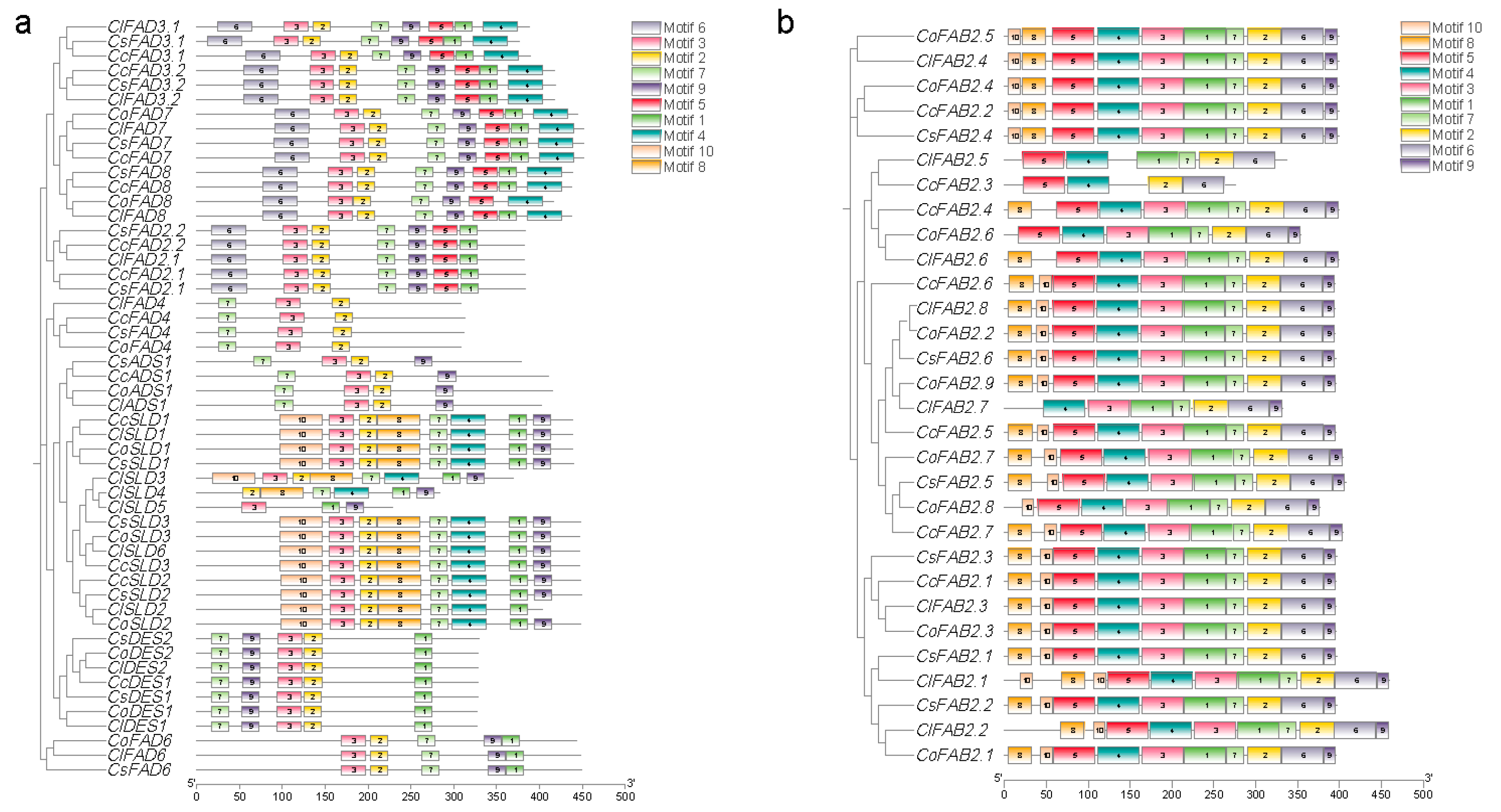
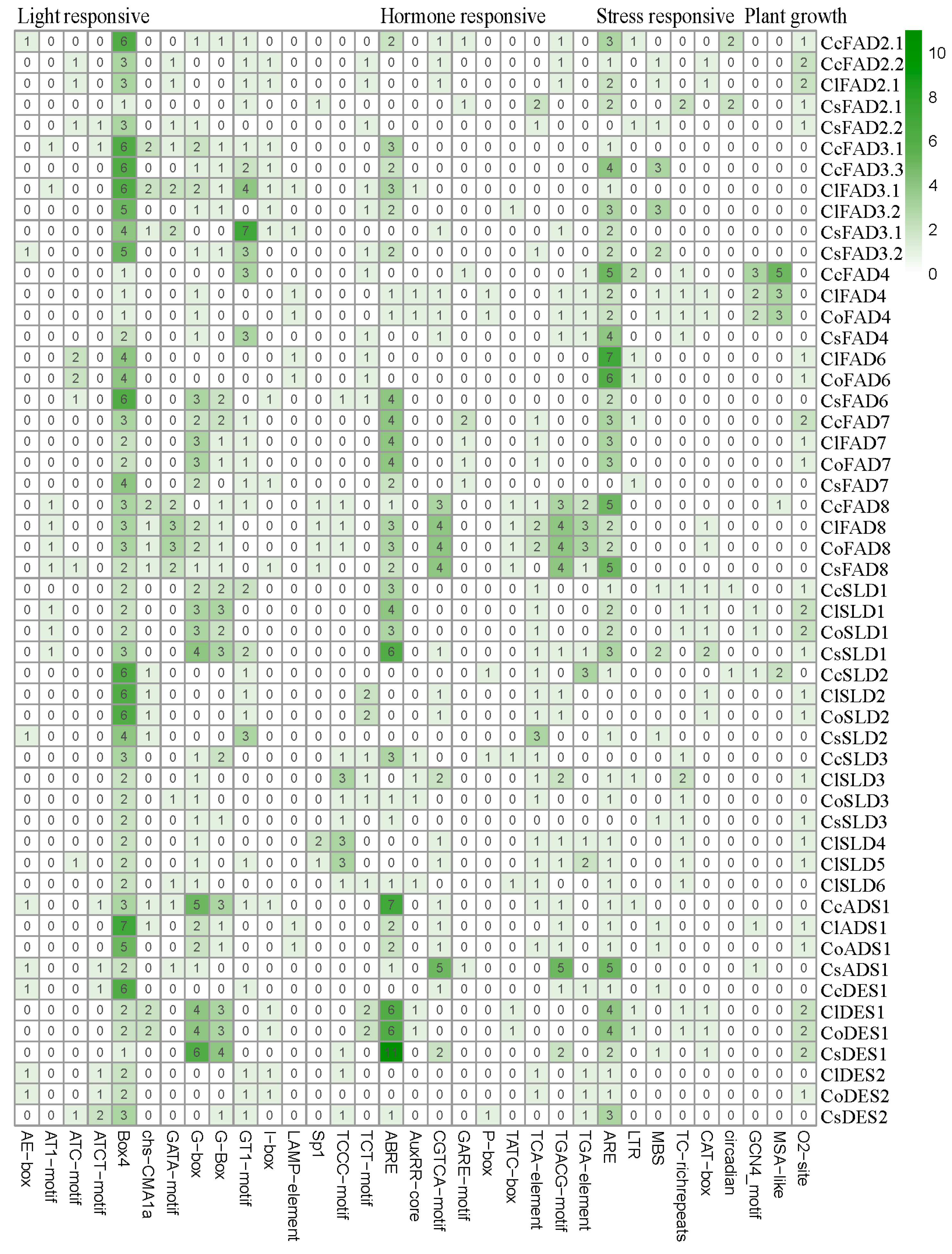
2.2. Identification and Bioinformatic Analysis of FAD Gene Family
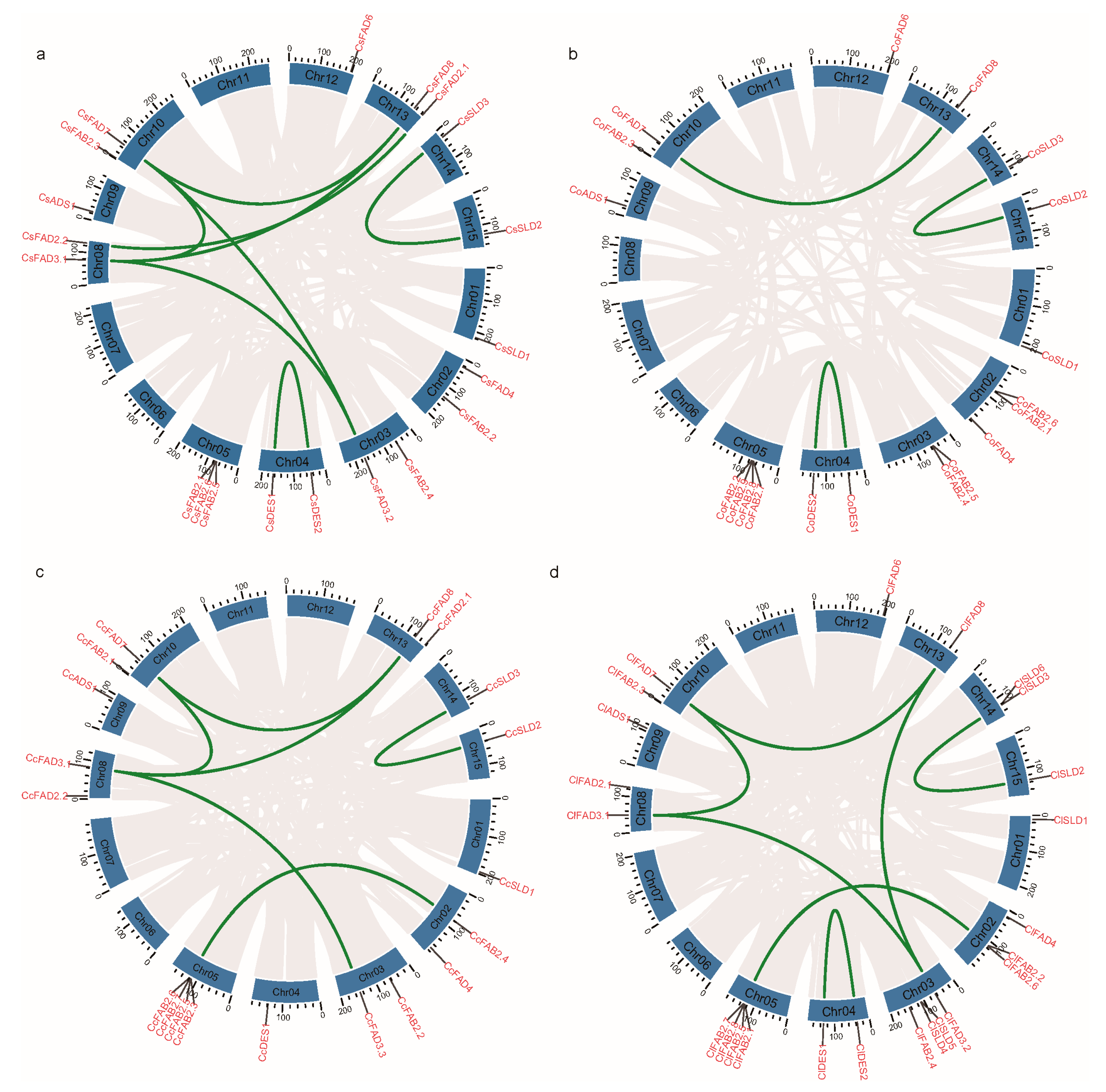
2.3. Chromosomal Location and Duplication Events of FAD Genes
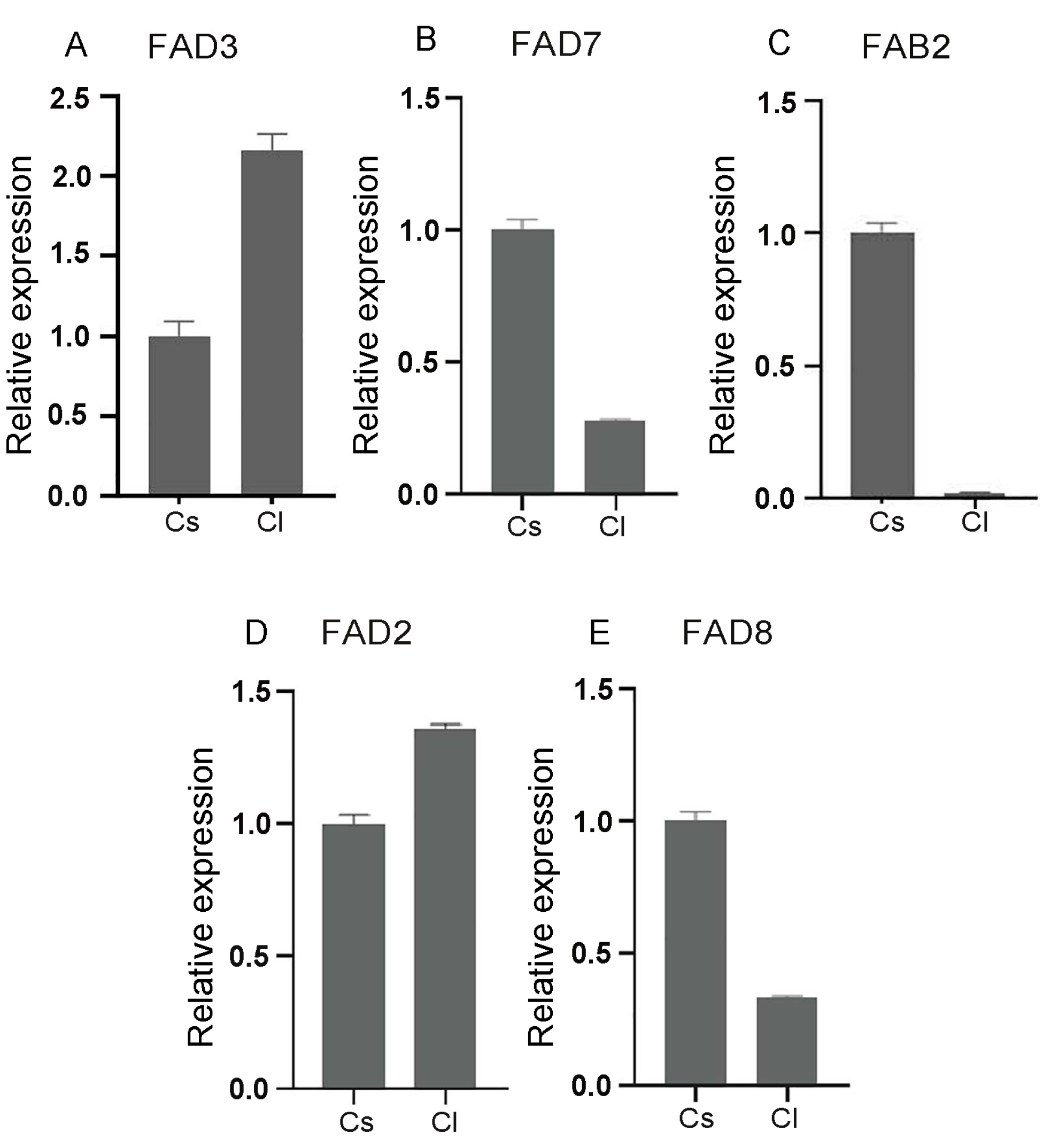
2.4. Comparative Expression Analysis of the FAD Gene Family in Seeds of Tea and Oil Tea
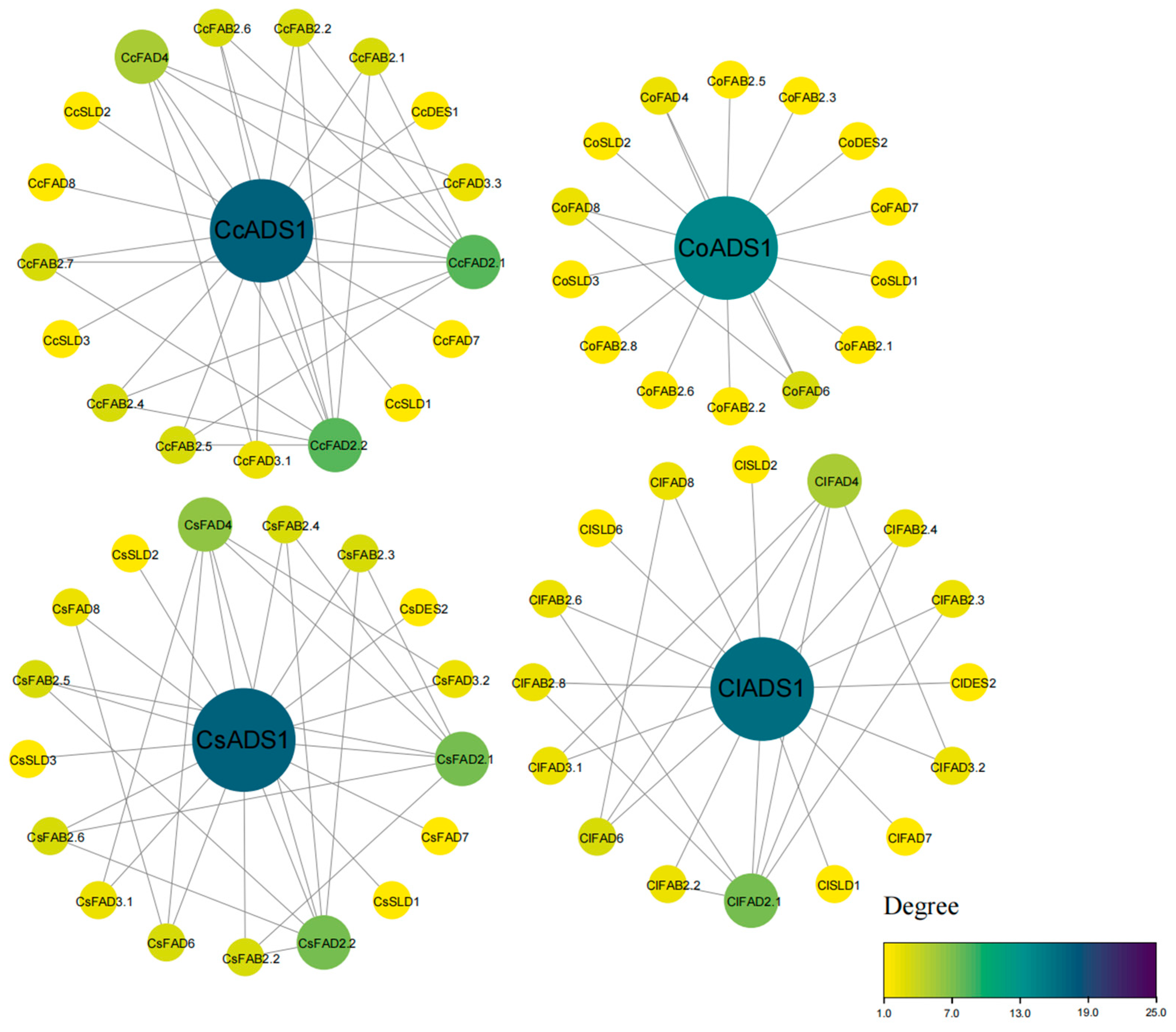
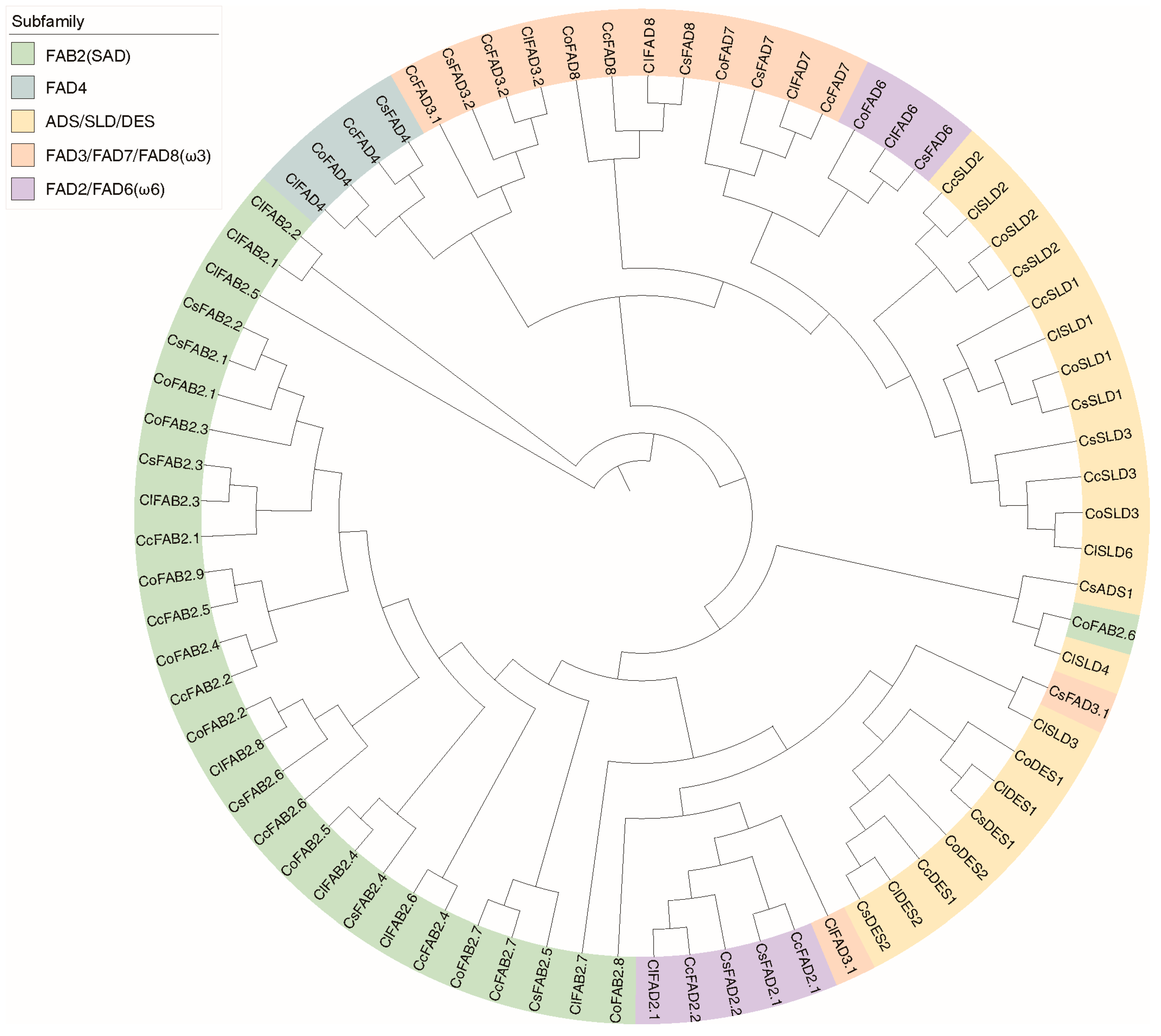
2.5. Clustering and Analysis of FAD Proteins in Tea and Oil Tea
3. Discussion
4. Materials and Methods
4.1. Genome Evolution and Expansion Analyses
4.2. Identification and Bioinformatic Analysis of FAD Gene Family
4.3. Chromosomal Location and Duplication Events of FAD Genes
4.4. Comparative Expression Analysis of the FAD Genes in Seeds of Tea and Oil Tea
4.5. Quantitative RT-PCR (qRT-PCR) in Seeds of Tea and Oil Tea
4.6. Protein Clustering and Analyzing
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vijayan, K.; Zhang, W.; Tsou, C. Molecular Taxonomy of Camellia (Theaceae) Inferred from nrITS Sequences. Am. J. Bot. 2009, 96, 1348–1360. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, Z.; Yang, Y. Genetic improvement and breeding of tea plant (Camellia sinensis) in China: From individual selection to hybridization and molecular breeding. Euphytica 2007, 154, 239–248. [Google Scholar] [CrossRef]
- Luan, F.; Zeng, J.; Yang, Y.; He, X.; Wang, B.; Gao, Y.; Zeng, N. Recent advances in Camellia oleifera Abel: A review of nutritional constituents, biofunctional properties, and potential industrial applications. J. Funct. Foods 2020, 275, 557–568. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, C.; Wen, Q.; Wang, N.; Tao, Y.; Xu, L. Deep sequencing of the Camellia chekiangoleosa transcriptome revealed candidate genes for anthocyanin biosynthesis. Gene 2014, 538, 1–7. [Google Scholar] [CrossRef]
- Tai, Y.; Wei, C.; Yang, H.; Zhang, L.; Chen, Q.; Deng, W.; Wei, S.; Zhang, J.; Fang, C.; Ho, C.; et al. Transcriptomic and phytochemical analysis of the biosynthesis of characteristic constituents in tea (Camellia sinensis) compared with oil tea (Camellia oleifera). BMC Plant Biol. 2015, 15, 190. [Google Scholar] [CrossRef]
- Chen, S.; Wang, P.; Kong, W.; Chai, K.; Zhang, S.; Yu, J.; Wang, Y.; Jiang, M.; Lei, W.; Chen, X.; et al. Gene mining and genomics-assisted breeding empowered by the pangenome of tea plant Camellia sinensis. Nat. Plants 2023, 9, 1986–1999. [Google Scholar] [CrossRef] [PubMed]
- Unver, T.; Wu, Z.; Sterck, L.; Turktas, M.; Lohaus, R.; Li, Z.; Yang, M.; He, L.; Deng, T.; Escalante, F.J.; et al. Genome of wild olive and the evolution of oil biosynthesis. Proc. Natl. Acad. Sci. USA 2017, 114, E9413–E9422. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Janila, P.; Pandey, M.K.; Shasidhar, Y.; Variath, M.T.; Sriswathi, M.; Khera, P.; Manohar, S.S.; Nagesh, P.; Vishwakarma, M.K.; Mishra, G.P.; et al. Molecular breeding for introgression of fatty acid desaturase mutant alleles (ahFAD2A and ahFAD2B) enhances oil quality in high and low oil containing peanut genotypes. Plant Sci. 2016, 242, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Wang, K.; Wang, Y.; Hu, Z.; Yan, C.; Huang, H.; Ma, X.; Cao, Y.; Long, W.; Liu, W.; et al. The genome of oil-Camellia and population genomics analysis provide insights into seed oil domestication. Genome Biol. 2022, 23, 14. [Google Scholar] [CrossRef]
- Shen, T.-F.; Huang, B.; Xu, M.; Zhou, P.-Y.; Ni, Z.-X.; Gong, C.; Wen, Q.; Cao, F.-L.; Xu, L.-A. The reference genome of Camellia chekiangoleosa provides insights into Camellia evolution and tea oil biosynthesis. Hortic. Res. 2022, 9, uhab083. [Google Scholar] [CrossRef]
- Gong, W.; Xiao, S.; Wang, L.; Liao, Z.; Chang, Y.; Mo, W.; Hu, G.; Li, W.; Zhao, G.; Zhu, H.; et al. Chromosome-level genome of Camellia lanceoleosa provides a valuable resource for understanding genome evolution and self-incompatibility. Plant J. 2022, 110, 881–898. [Google Scholar] [CrossRef]
- Saini, R.; Kumar, S. Genome-wide identification, characterization and in-silico profiling of genes encoding FAD (fatty acid desaturase) proteins in chickpea (Cicer arietinum, L.). Plant Gene 2019, 18, 100180. [Google Scholar] [CrossRef]
- Heppard, E.P.; Kinney, A.J.; Stecca, K.L.; Miao, G.H. Developmental and growth temperature regulation of two different microsomal omega-6 desaturase genes in soybeans. Plant Physiol. 1996, 110, 311–319. [Google Scholar] [CrossRef]
- Jung, S.; Swift, D.; Sengoku, E.; Patel, M.; Teulé, F.; Powell, G.; Moore, K.; Abbott, A. The high oleate trait in the cultivated peanut [Arachis hypogaea, L.]. I. Isolation and characterization of two genes encoding microsomal oleoyl-PC desaturases. Mol. Gen. Genet. 2000, 263, 796–805. [Google Scholar] [CrossRef]
- Dyer, J.M.; Chapital, D.C.; Kuan, J.-C.W.; Mullen, R.T.; Turner, C.; McKeon, T.A.; Pepperman, A.B. Molecular Analysis of a Bifunctional Fatty Acid Conjugase/Desaturase from Tung. Implications for the Evolution of Plant Fatty Acid Diversity. Plant Physiol. 2002, 130, 2027–2038. [Google Scholar] [CrossRef]
- Zhang, B.; Xia, P.; Yu, H.; Li, W.; Chai, W.; Liang, Z. Based on the whole genome clarified the evolution and expression process of fatty acid desaturase genes in three soybeans. Int. J. Biol. Macromol. 2021, 182, 1966–1980. [Google Scholar] [CrossRef]
- Maeda, H.; Sage, T.L.; Isaac, G.; Welti, R.; DellaPenna, D. Tocopherols Modulate Extraplastidic Polyunsaturated Fatty Acid Metabolism in Arabidopsis at Low Temperature. Plant Cell 2008, 20, 452–470. [Google Scholar] [CrossRef]
- Cheng, C.; Liu, F.; Sun, X.; Wang, B.; Liu, J.; Ni, X.; Hu, C.; Deng, G.; Tong, Z.; Zhang, Y.; et al. Genome-wide identification of FAD gene family and their contributions to the temperature stresses and mutualistic and parasitic fungi colonization responses in banana. Int. J. Biol. Macromol. 2022, 204, 661–676. [Google Scholar] [CrossRef]
- Chen, C.; Yang, J.; Tong, H.; Li, T.; Wang, L.; Chen, H. Genome-wide analysis of fatty acid desaturase genes in rice (Oryza sativa, L.). Sci. Rep. 2019, 9, 19445. [Google Scholar]
- Xue, Y.; Chen, B.; Wang, R.; Win, A.N.; Li, J.; Chai, Y. Genome-Wide Survey and Characterization of Fatty Acid Desaturase Gene Family in Brassica napus and Its Parental Species. Appl. Biochem. Biotechnol. 2018, 184, 582–598. [Google Scholar] [CrossRef]
- Hajiahmadi, Z.; Abedi, A.; Wei, H.; Sun, W.; Ruan, H.; Zhuge, Q.; Movahedi, A. Identification, evolution, expression, and docking studies of fatty acid desaturase genes in wheat (Triticum aestivum, L.). BMC Genom. 2020, 21, 778. [Google Scholar] [CrossRef]
- Ohlrogge, J.; Browse, J. Lipid biosynthesis. Plant Cell 1995, 7, 957–970. [Google Scholar]
- Dar, A.A.; Choudhury, A.R.; Kancharla, P.K.; Arumugam, N. The FAD2 Gene in Plants: Occurrence, Regulation, and Role. Front. Plant Sci. 2017, 8, 1789. [Google Scholar] [CrossRef]
- Li, K.R.; Qi, L.L.; Zhao, S.D. A study of the physical and chemical properties of the seed oil and its fatty acid compositions of 27 species of Camellia. J. Cent.-S. For. Coll. 1984, 02, 101–109. [Google Scholar]
- Wang, Y.Y.; Song, X.H.; Li, C.R. Fatty acids analysis of seeds oil from eight kinds of Camellia plants. China Oils Fats 2007, 09, 78–79. [Google Scholar]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Marks, D.S.; Hopf, T.A.; Sander, C. Protein structure prediction from sequence variation. Nat. Biotechnol. 2012, 30, 1072–1080. [Google Scholar] [CrossRef]
- Moore, A.D.; Björklund, Å.K.; Ekman, D.; Bornberg-Bauer, E.; Elofsson, A. Arrangements in the modular evolution of proteins. Trends Biochem. Sci. 2008, 33, 444–451. [Google Scholar] [CrossRef]
- Mittal, S.; Saluja, D. Protein Post-translational Modifications: Role in Protein Structure, Function and Stability. In Proteostasis and Chaperone Surveillance; Singh, L.R., Dar, T.A., Ahmad, P., Eds.; Springer: New Delhi, India, 2015; pp. 25–37. [Google Scholar]
- Wang, X.; Zeng, Q.; del Mar Contreras, M.; Wang, L. Profiling and quantification of phenolic compounds in Camellia seed oils: Natural tea polyphenols in vegetable oil. Food Res. Int. 2017, 102, 184–194. [Google Scholar] [CrossRef]
- Zeng, W.; Endo, Y. Lipid Characteristics of Camellia Seed Oil. J. Oleo Sci. 2019, 68, 649–658. [Google Scholar] [CrossRef]
- Wallis, J.G.; Browse, J. Mutants of Arabidopsis reveal many roles for membrane lipids. Prog. Lipid Res. 2002, 41, 254–278. [Google Scholar] [CrossRef]
- Chi, X.; Yang, Q.; Lu, Y.; Wang, J.; Zhang, Q.; Pan, L.; Chen, M.; He, Y.; Yu, S. Genome-Wide Analysis of Fatty Acid Desaturases in Soybean (Glycine max). Plant Mol. Biol. Report. 2011, 29, 769–783. [Google Scholar] [CrossRef]
- Chi, X.; Yang, Q.; Pan, L.; Chen, M.; He, Y.; Yang, Z.; Yu, S. Isolation and characterization of fatty acid desaturase genes from peanut (Arachis hypogaea, L.). Plant Cell Rep. 2011, 30, 1393–1404. [Google Scholar] [CrossRef]
- Itoh M, Nacher JC, Kuma K-i, Goto S, Kanehisa M: Evolutionary history and functional implications of protein domains and their combinations in eukaryotes. Genome Biol. 2007, 8, R121.
- Jiaying, H.; Qiupeng, L.; Hongyuan, F.; Zixin, H.; Hu, X.; Yunjia, L.; Kunli, Q.; Peng, H.; Qiang, G.; Boshu, L.; et al. Discovery of deaminase functions by structure-based protein clustering. Cell 2023, 186, 3182–3195. [Google Scholar]
- Jiao, P.; Wang, B.; Wang, X.; Liu, B.; Wang, Y.; Li, J. Struct2GO: Protein function prediction based on graph pooling algorithm and AlphaFold2 structure information. Bioinformatics 2023, 39, btad637. [Google Scholar] [CrossRef]
- Sun, J.; Lu, F.; Luo, Y.; Bie, L.; Xu, L.; Wang, Y. OrthoVenn3: An integrated platform for exploring and visualizing orthologous data across genomes. Nucleic Acids Res. 2023, 51, W397–W403. [Google Scholar] [CrossRef]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 2020, 36, 5516–5518. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2023, 2, e107. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Miao, B.; Dong, W.; Han, Z.; Luo, X.; Ke, C.; You, W. TOmicsVis: An all-in-one transcriptomic analysis and visualization R package with Shinyapp interface. iMeta 2023, 2, e137. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Z.; Mao, D.; Wang, Y.; Deng, H.; Liu, X.; Zhang, T.; Han, Z.; Zhang, X. Comparative Genome-Wide Identification of the Fatty Acid Desaturase Gene Family in Tea and Oil Tea. Plants 2024, 13, 1444. https://doi.org/10.3390/plants13111444
Ye Z, Mao D, Wang Y, Deng H, Liu X, Zhang T, Han Z, Zhang X. Comparative Genome-Wide Identification of the Fatty Acid Desaturase Gene Family in Tea and Oil Tea. Plants. 2024; 13(11):1444. https://doi.org/10.3390/plants13111444
Chicago/Turabian StyleYe, Ziqi, Dan Mao, Yujian Wang, Hongda Deng, Xing Liu, Tongyue Zhang, Zhiqiang Han, and Xingtan Zhang. 2024. "Comparative Genome-Wide Identification of the Fatty Acid Desaturase Gene Family in Tea and Oil Tea" Plants 13, no. 11: 1444. https://doi.org/10.3390/plants13111444
APA StyleYe, Z., Mao, D., Wang, Y., Deng, H., Liu, X., Zhang, T., Han, Z., & Zhang, X. (2024). Comparative Genome-Wide Identification of the Fatty Acid Desaturase Gene Family in Tea and Oil Tea. Plants, 13(11), 1444. https://doi.org/10.3390/plants13111444