

Genomic and Transcriptomic Analysis of Pea (Pisum sativum L.) Breeding Line ‘Triumph’ with High Symbiotic Responsivity

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
2.1. Genome Sequencing of the Breeding Line ‘Triumph’ and Its Parental Cultivars
2.1.1. Sequencing and Data Processing
2.1.2. Portion of the ‘Triumph’s’ Genome Inherited from cv. ‘Vendevil’
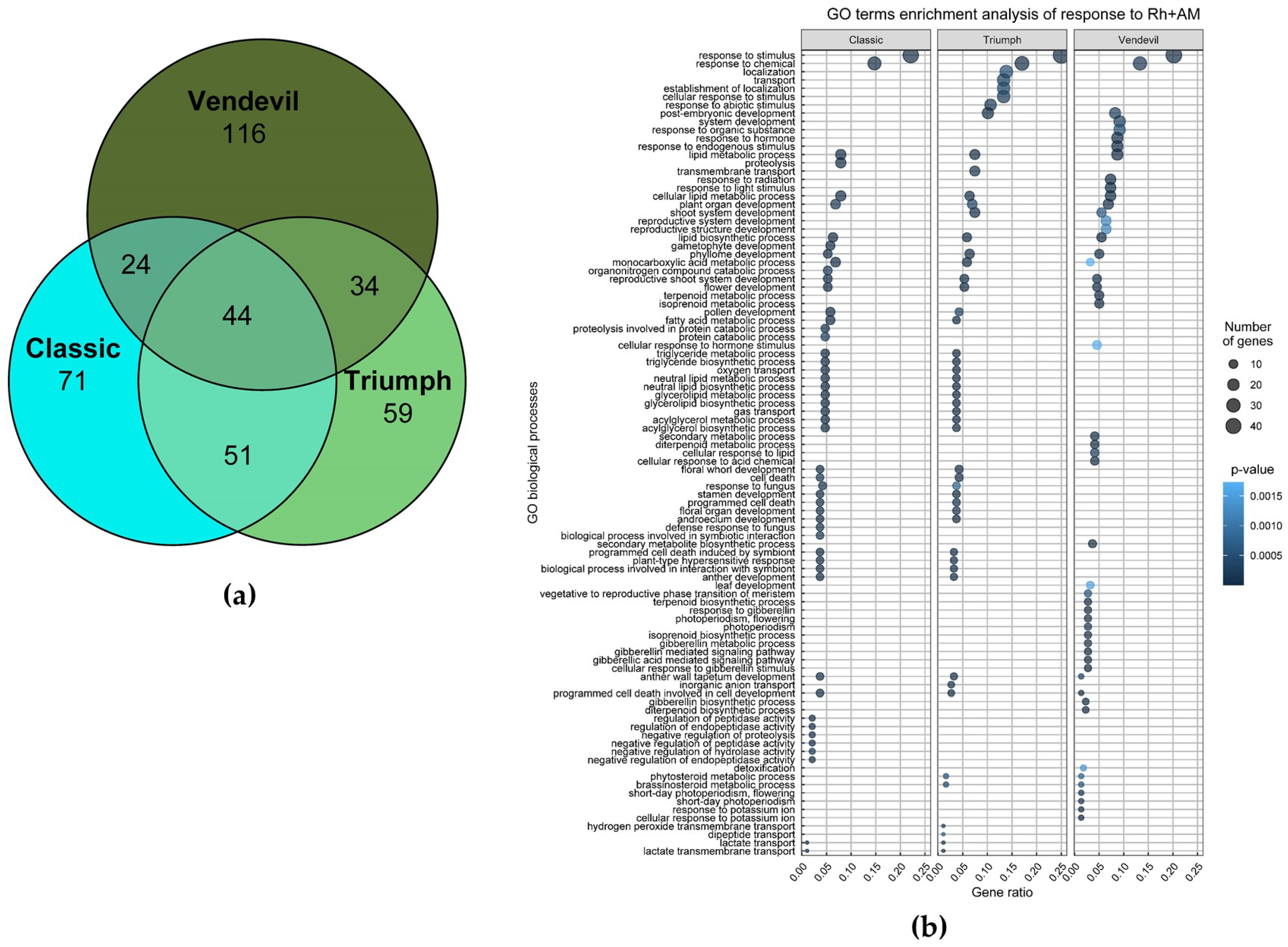
2.2. Transcriptomic Analysis of Responses of ‘Triumph’, ‘Vendevil’ and ‘Classic’ to Inoculation with Rhizobia and AM Fungi
2.2.1. The Effect of Combined Inoculation with Nodule Bacteria and AM Fungi on Growth Parameters of the Studied Pea Genotypes
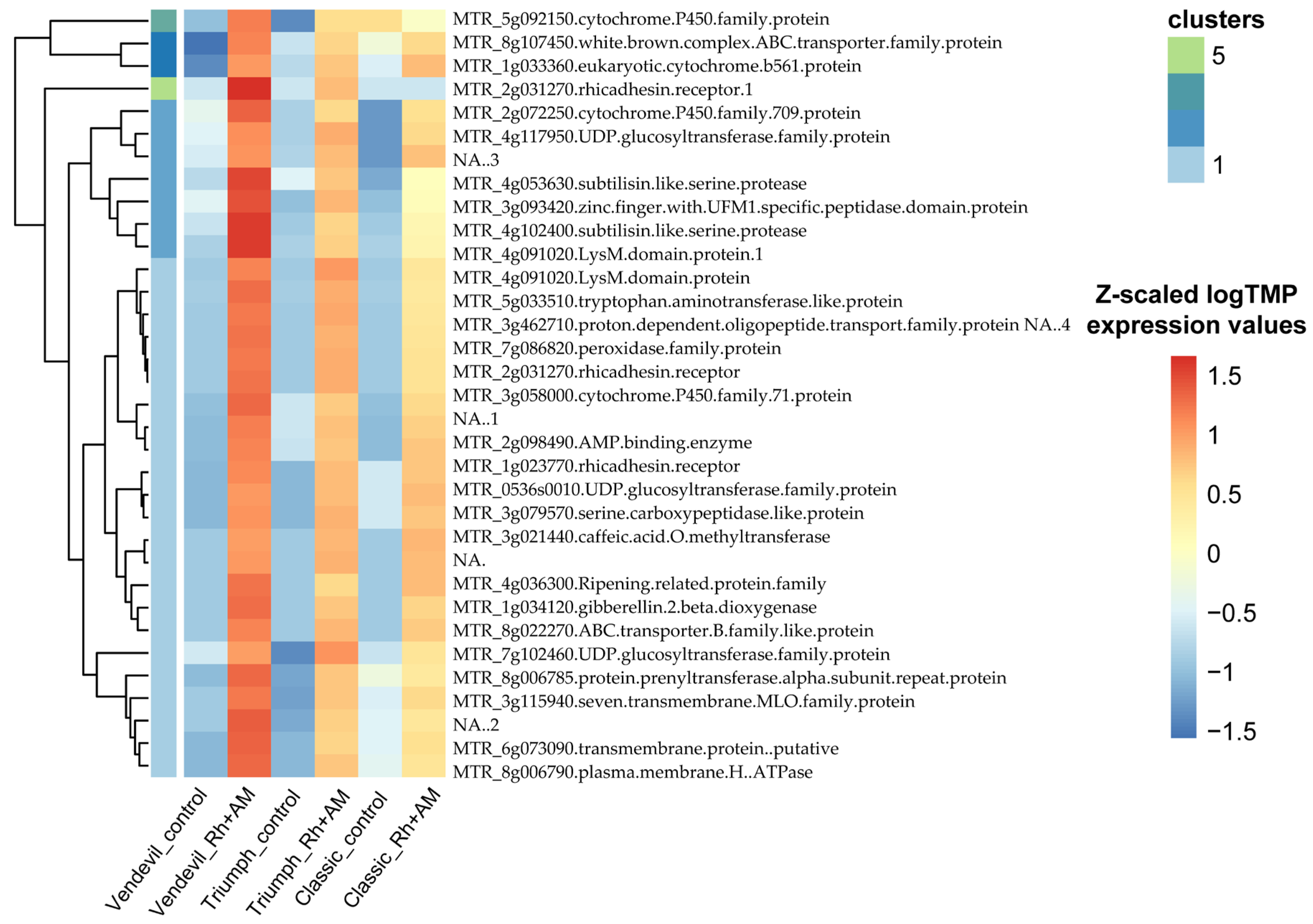
2.2.2. Transcriptomic Analysis of the Response to Inoculation
2.2.3. Allele-Specific Gene Expression in Shoots and Roots of ‘Triumph’
2.2.4. qRT-PCR Validation of Transcriptomic Data
3. Discussion
4. Materials and Methods
4.1. Plants Material and Growth Condition
Experimental Setup for the Pot Experiment
4.2. Statistical Analysis of Plant Growth and Yield Parameters
4.3. Nuclei and DNA Isolation
4.3.1. Isolation of P. sativum Nuclei
4.3.2. Isolation of Pea Nuclear DNA
4.4. Whole Genome Sequencing and Reads Processing
4.5. SNV Calling and Identification of Genome Regions Inherited by ‘Triumph’ from ‘Vendevil’
4.6. Functional Analysis of Genes
4.7. RNA Isolation
4.8. Transcriptome Reads Processing
4.9. Differential Expression Analysis
4.10. Phylogenetic Analysis
4.11. Real-Time qRT-PCR
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, D.; Dong, W.; Murray, J.; Wang, E. Innovation and Appropriation in Mycorrhizal and Rhizobial Symbioses. Plant Cell 2022, 34, 1573–1599. [Google Scholar] [CrossRef]
- Dilworth, M.J.; James, E.K.; Sprent, J.I.; Newton, W.E. Nitrogen-Fixing Leguminous Symbioses; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2008; Volume 7, ISBN 1402035489. [Google Scholar]
- Liu, A.; Ku, Y.S.; Contador, C.A.; Lam, H.M. The Impacts of Domestication and Agricultural Practices on Legume Nutrient Acquisition through Symbiosis with Rhizobia and Arbuscular Mycorrhizal Fungi. Front. Genet. 2020, 11, 583954. [Google Scholar] [CrossRef]
- Peoples, M.B.; Herridge, D.F.; Ladha, J.K. Biological Nitrogen Fixation: An Efficient Source of Nitrogen for Sustainable Agricultural Production? Plant Soil 1995, 174, 3–28. [Google Scholar] [CrossRef]
- Zahran, H.H. Rhizobium-Legume Symbiosis and Nitrogen Fixation under Severe Conditions and in an Arid Climate. Microbiol. Mol. Biol. Rev. 1999, 63, 968–989. [Google Scholar] [CrossRef]
- Clúa, J.; Roda, C.; Zanetti, M.E.; Blanco, F.A. Compatibility between Legumes and Rhizobia for the Establishment of a Successful Nitrogen-Fixing Symbiosis. Genes 2018, 9, 125. [Google Scholar] [CrossRef]
- Fall, A.F.; Nakabonge, G.; Ssekandi, J.; Founoune-Mboup, H.; Apori, S.O.; Ndiaye, A.; Badji, A.; Ngom, K. Roles of Arbuscular Mycorrhizal Fungi on Soil Fertility: Contribution in the Improvement of Physical, Chemical, and Biological Properties of the Soil. Front. Fungal Biol. 2022, 3, 723892. [Google Scholar] [CrossRef]
- Shtark, O.Y.; Zhukov, V.A.; Sulima, A.S.; Singh, R.; Naumkina, T.S.; Borisov, A.Y. Prospects for the Use of Multi-Component Symbiotic Systems of the Legumes. Ecol. Genet. 2015, 13, 33–46. [Google Scholar] [CrossRef]
- Roy, S.; Liu, W.; Nandety, R.S.; Crook, A.; Mysore, K.S.; Pislariu, C.I.; Frugoli, J.; Dickstein, R.; Udvardi, M.K. Celebrating 20 Years of Genetic Discoveries in Legume Nodulation and Symbiotic Nitrogen Fixation. Plant Cell 2020, 32, 15–41. [Google Scholar] [CrossRef]
- Bourion, V.; Heulin-Gotty, K.; Aubert, V.; Tisseyre, P.; Chabert-Martinello, M.; Pervent, M.; Delaitre, C.; Vile, D.; Siol, M.; Duc, G.; et al. Co-Inoculation of a Pea Core-Collection with Diverse Rhizobial Strains Shows Competitiveness for Nodulation and Efficiency of Nitrogen Fixation Are Distinct Traits in the Interaction. Front. Plant Sci. 2018, 8, 2249. [Google Scholar] [CrossRef]
- Lindström, K.; Mousavi, S.A. Effectiveness of Nitrogen Fixation in Rhizobia. Microb. Biotechnol. 2020, 13, 1314–1335. [Google Scholar] [CrossRef]
- Van Der Heijden, M.G.A. Arbuscular Mycorrhizal Fungi as a Determinant of Plant Diversity: In Search of Underlying Mechanisms and General Principles. In Mycorrhizal Ecology; Springer: Berlin/Heidelberg, Germany, 2002; pp. 243–265. [Google Scholar]
- Pandey, A.K.; Rubiales, D.; Wang, Y.; Fang, P.; Sun, T.; Liu, N.; Xu, P. Omics Resources and Omics-Enabled Approaches for Achieving High Productivity and Improved Quality in Pea (Pisum Sativum L.). Theor. Appl. Genet. 2021, 134, 755–776. [Google Scholar] [CrossRef]
- Bagheri, M.; Santos, C.S.; Rubiales, D.; Vasconcelos, M.W. Challenges in Pea Breeding for Tolerance to Drought: Status and Prospects. Ann. Appl. Biol. 2023, 183, 108–120. [Google Scholar] [CrossRef]
- Rubiales, D.; Barilli, E.; Rispail, N. Breeding for Biotic Stress Resistance in Pea. Agriculture 2023, 13, 1825. [Google Scholar] [CrossRef]
- Shtark, O.Y.; Borisov, A.Y.; Zhukov, V.A.; Tikhonovich, I.A. Mutually Beneficial Legume Symbioses with Soil Microbes and Their Potential for Plant Production. Symbiosis 2012, 58, 51–62. [Google Scholar] [CrossRef]
- Yakobi, L.M.; Kukalev, A.S.; Ushakov, K.V.; Tsyganov, V.E.; Naumkina, T.S.; Provorov, N.A.; Borisov, A.Y.; Tikhonovich, I.A. Polymorphism of Garden Pea Forms by the Effectiveness of Symbiosis with the Endomycorrhizal Fungus Glomus Sp. under Conditions of Inoculation with Rhizobia. Agric. Biol. Sel’skokhozyaistvennaya Biol. 2000, 2000, 94–102. [Google Scholar]
- Zhukov, V.A.; Zhernakov, A.I.; Sulima, A.S.; Kulaeva, O.A.; Kliukova, M.S.; Afonin, A.M.; Shtark, O.Y.; Tikhonovich, I.A. Association Study of Symbiotic Genes in Pea (Pisum Sativum L.) Cultivars Grown in Symbiotic Conditions. Agronomy 2021, 11, 2368. [Google Scholar] [CrossRef]
- Naumkina, T.S. Breeding Peas (Pisum Sativum L.) to Increase the Efficiency of Symbiotic Nitrogen Fixation, Diss. Dr. Sci. State Scientific Institution All-Russian Research Institute of Grain Legumes and Cereals of the Russian Agricultural Academy. 2007. Available online: https://www.dissercat.com/content/selektsiya-gorokha-pisum-sativum-l-na-povyshenie-effektivnosti-simbioticheskoi-azotfiksatsii/read (accessed on 12 December 2023).
- Shtark, O.Y.; Danilova, T.N.; Naumkina, T.S.; Vasilchikov, A.G.; Chebotar, V.K.; Kazakov, A.E.; Zhernakov, A.I.; Nemankin, T.A.; Prilepskaya, N.A.; Borisov, A.U. Analysis of Pea (Pisum Sativum L.) Source Material for Breeding of Cultivars with High Symbiotic Potential and Choice of Criteria for Its Evaluation. Ecol. Genet. 2006, 4, 22–28. [Google Scholar] [CrossRef]
- Zorin, E.A.; Kliukova, M.S.; Afonin, A.M.; Gribchenko, E.S.; Gordon, M.L.; Sulima, A.S.; Zhernakov, A.I.; Kulaeva, O.A.; Romanyuk, D.A.; Kusakin, P.G.; et al. A Variable Gene Family Encoding Nodule-Specific Cysteine-Rich Peptides in Pea (Pisum Sativum L.). Front. Plant Sci. 2022, 13, 884726. [Google Scholar] [CrossRef]
- Kaiser, B.N.; Moreau, S.; Castelli, J.; Thomson, R.; Lambert, A.; Bogliolo, S.; Puppo, A.; Day, D.A. The Soybean NRAMP Homologue, GmDMT1, Is a Symbiotic Divalent Metal Transporter Capable of Ferrous Iron Transport. Plant J. 2003, 35, 295–304. [Google Scholar] [CrossRef]
- Middleton, P.H.; Jakab, J.; Penmetsa, R.V.; Starker, C.G.; Doll, J.; Kalo, P.; Prabhu, R.; Marsh, J.F.; Mitra, R.M.; Kereszt, A.; et al. An ERF Transcription Factor in Medicago truncatula That Is Essential for Nod Factor Signal Transduction. Plant Cell Online 2007, 19, 1221–1234. [Google Scholar] [CrossRef]
- Wu, F.-Q.; Zhang, X.-M.; Li, D.-M.; Fu, Y.-F. Ectopic Expression Reveals a Conserved PHYB Homolog in Soybean. PLoS ONE 2011, 6, e27737. [Google Scholar] [CrossRef]
- Haney, C.H.; Riely, B.K.; Tricoli, D.M.; Cook, D.R.; Ehrhardt, D.W.; Long, S.R. Symbiotic Rhizobia Bacteria Trigger a Change in Localization and Dynamics of the Medicago truncatula Receptor Kinase LYK3. Plant Cell 2011, 23, 2774–2787. [Google Scholar] [CrossRef]
- Mbengue, M.; Camut, S.; de Carvalho-Niebel, F.; Deslandes, L.; Froidure, S.; Klaus-Heisen, D.; Moreau, S.; Rivas, S.; Timmers, T.; Hervé, C.; et al. The Medicago truncatula E3 Ubiquitin Ligase PUB1 Interacts with the LYK3 Symbiotic Receptor and Negatively Regulates Infection and Nodulation. Plant Cell 2010, 22, 3474–3488. [Google Scholar] [CrossRef]
- Jia, H.; Ren, H.; Gu, M.; Zhao, J.; Sun, S.; Zhang, X.; Chen, J.; Wu, P.; Xu, G. The Phosphate Transporter Gene Ospht1;8 Is Involved in Phosphate Homeostasis in Rice. Plant Physiol. 2011, 156, 1164–1175. [Google Scholar] [CrossRef]
- Breuillin-Sessoms, F.; Floss, D.S.; Karen Gomez, S.; Pumplin, N.; Ding, Y.; Levesque-Tremblay, V.; Noar, R.D.; Daniels, D.A.; Bravo, A.; Eaglesham, J.B.; et al. Suppression of Arbuscule Degeneration in Medicago truncatula Phosphate Transporter4 Mutants Is Dependent on the Ammonium Transporter 2 Family Protein AMT2;3. Plant Cell 2015, 27, 1352–1366. [Google Scholar] [CrossRef]
- Gutjahr, C.; Radovanovic, D.; Geoffroy, J.; Zhang, Q.; Siegler, H.; Chiapello, M.; Casieri, L.; An, K.; An, G.; Guiderdoni, E. The Half-size ABC Transporters STR1 and STR2 Are Indispensable for Mycorrhizal Arbuscule Formation in Rice. Plant J. 2012, 69, 906–920. [Google Scholar] [CrossRef]
- Zhang, Q.; Blaylock, L.A.; Harrison, M.J. Two Medicago Truncatula Half-ABC Transporters Are Essential for Arbuscule Development in Arbuscular Mycorrhizal Symbiosis. Plant Cell 2010, 22, 1483–1497. [Google Scholar] [CrossRef]
- Wallace, I.S.; Choi, W.G.; Roberts, D.M. The Structure, Function and Regulation of the Nodulin 26-like Intrinsic Protein Family of Plant Aquaglyceroporins. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1165–1175. [Google Scholar] [CrossRef]
- Parker, J.L.; Newstead, S. Molecular Basis of Nitrate Uptake by the Plant Nitrate Transporter NRT1.1. Nature 2014, 507, 68–72. [Google Scholar] [CrossRef]
- Mohammadi-Dehcheshmeh, M.; Niazi, A.; Ebrahimi, M.; Tahsili, M.; Nurollah, Z.; Ebrahimi Khaksefid, R.; Ebrahimi, M.; Ebrahimie, E. Unified Transcriptomic Signature of Arbuscular Mycorrhiza Colonization in Roots of Medicago truncatula by Integration of Machine Learning, Promoter Analysis, and Direct Merging Meta-Analysis. Front. Plant Sci. 2018, 871, 1550. [Google Scholar] [CrossRef]
- Schaller, F.; Biesgen, C.; Müssig, C.; Altmann, T.; Weiler, E.W. 12-Oxophytodienoate Reductase 3 (OPR3) Is the Isoenzyme Involved in Jasmonate Biosynthesis. Planta 2000, 210, 979–984. [Google Scholar] [CrossRef]
- Goyal, R.K.; Mattoo, A.K.; Schmidt, M.A. Rhizobial–Host Interactions and Symbiotic Nitrogen Fixation in Legume Crops Toward Agriculture Sustainability. Front. Microbiol. 2021, 12, 669404. [Google Scholar] [CrossRef]
- Sinjushin, A.; Semenova, E.; Vishnyakova, M. Usage of Morphological Mutations for Improvement of a Garden Pea (Pisum sativum): The Experience of Breeding in Russia. Agronomy 2022, 12, 544. [Google Scholar] [CrossRef]
- Kuzmicheva, Y.V.; Shaposhnikov, A.I.; Azarova, T.S.; Petrova, S.N.; Naumkina, T.S.; Borisov, A.Y.; Belimov, A.A.; Kravchenko, L.V.; Parakhin, N.V.; Tikhonovich, I.A. Composition of Root Exometabolites of the Symbiotically Effective Pea Cultivar Triumph and Its Parental Forms. Russ. J. Plant Physiol. 2014, 61, 112–118. [Google Scholar] [CrossRef]
- Hedden, P.; Thomas, S.G. Gibberellin Biosynthesis and Its Regulation. Biochem. J. 2012, 444, 11–25. [Google Scholar] [CrossRef]
- Lange, T.; Lange, M.J.P. The Multifunctional Dioxygenases of Gibberellin Synthesis. Plant Cell Physiol. 2020, 61, 1869–1879. [Google Scholar] [CrossRef]
- Kawai, Y.; Ono, E.; Mizutani, M. Evolution and Diversity of the 2-Oxoglutarate-Dependent Dioxygenase Superfamily in Plants. Plant J. 2014, 78, 328–343. [Google Scholar] [CrossRef]
- McAdam, E.L.; Reid, J.B.; Foo, E. Gibberellins Promote Nodule Organogenesis but Inhibit the Infection Stages of Nodulation. J. Exp. Bot. 2018, 69, 2117–2130. [Google Scholar] [CrossRef]
- Igielski, R.; Kępczyńska, E. Gene Expression and Metabolite Profiling of Gibberellin Biosynthesis during Induction of Somatic Embryogenesis in Medicago truncatula Gaertn. PLoS ONE 2017, 12, 182055. [Google Scholar] [CrossRef]
- Afonin, A.M.; Gribchenko, E.S.; Zorin, E.A.; Sulima, A.S.; Romanyuk, D.A.; Zhernakov, A.I.; Shtark, O.Y.; Akhtemova, G.A.; Zhukov, V.A. Unique Transcriptome Features of Pea (Pisum sativum L.) Lines with Differing Responses to Beneficial Soil Microorganisms. Ecol. Genet. 2021, 19, 131–141. [Google Scholar] [CrossRef]
- Ku, Y.S.; Contador, C.A.; Ng, M.S.; Yu, J.; Chung, G.; Lam, H.M. The Effects of Domestication on Secondary Metabolite Composition in Legumes. Front. Genet. 2020, 11, 581357. [Google Scholar] [CrossRef] [PubMed]
- Tawaraya, K. Arbuscular Mycorrhizal Dependency of Different Plant Species and Cultivars. Soil Sci. Plant Nutr. 2003, 49, 655–668. [Google Scholar] [CrossRef]
- McGuiness, P.N.; Reid, J.B.; Foo, E. The Role of Gibberellins and Brassinosteroids in Nodulation and Arbuscular Mycorrhizal Associations. Front. Plant Sci. 2019, 10, 269. [Google Scholar] [CrossRef] [PubMed]
- Foo, E.; Ross, J.J.; Jones, W.T.; Reid, J.B. Plant Hormones in Arbuscular Mycorrhizal Symbioses: An Emerging Role for Gibberellins. Ann. Bot. 2013, 111, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Schaller, F. Enzymes of the Biosynthesis of Octadecanoid-derived Signalling Molecules. J. Exp. Bot. 2001, 52, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Dunwell, J.M.; Gibbings, J.G.; Mahmood, T.; Saqlan Naqvi, S.M. Germin and Germin-like Proteins: Evolution, Structure, and Function. CRC. Crit. Rev. Plant Sci. 2008, 27, 342–375. [Google Scholar] [CrossRef]
- Barman, A.R.; Banerjee, J. Versatility of Germin-like Proteins in Their Sequences, Expressions, and Functions. Funct. Integr. Genomics 2015, 15, 533–548. [Google Scholar] [CrossRef]
- Gucciardo, S.; Wisniewski, J.P.; Brewin, N.J.; Bornemann, S. A Germin-like Protein with Superoxide Dismutase Activity in Pea Nodules with High Protein Sequence Identity to a Putative Rhicadhesin Receptor. J. Exp. Bot. 2007, 58, 1161–1171. [Google Scholar] [CrossRef]
- Doll, J.; Hause, B.; Demchenko, K.; Pawlowski, K.; Krajinski, F. A Member of the Germin-Like Protein Family Is a Highly Conserved Mycorrhiza-Specific Induced Gene. Plant Cell Physiol. 2003, 44, 1208–1214. [Google Scholar] [CrossRef][Green Version]
- Chakraborty, S.; Driscoll, H.E.; Abrahante, J.E.; Zhang, F.; Fisher, R.F.; Harris, J.M. Salt Stress Enhances Early Symbiotic Gene Expression in Medicago truncatula and Induces a Stress-Specific Set of Rhizobium-Responsive Genes. Mol. Plant-Microbe Interact. 2021, 34, 904–921. [Google Scholar] [CrossRef]
- Afonin, A.; Sulima, A.; Zhernakov, A.; Zhukov, V. Draft Genome of the Strain RCAM1026 Rhizobium leguminosarum Bv. Viciae. Genomics Data 2017, 11, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Sikorskaite, S.; Rajamäki, M.L.; Baniulis, D.; Stanys, V.; Valkonen, J.P.T. Protocol: Optimised Methodology for Isolation of Nuclei from Leaves of Species in the Solanaceae and Rosaceae Families. Plant Methods 2013, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Zerpa-Catanho, D.; Zhang, X.; Song, J.; Hernandez, A.G.; Ming, R. Ultra-Long DNA Molecule Isolation from Plant Nuclei for Ultra-Long Read Genome Sequencing. STAR Protoc. 2021, 2, 100343. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.O.; Bendich, A.J. Extraction of DNA from Milligram Amounts of Fresh, Herbarium and Mummified Plant Tissues. Plant Mol. Biol. 1985, 5, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab (LBNL): Berkeley, CA, USA, 2014; Volume 13. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila melanogaster Strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Bolger, M.; Schwacke, R.; Usadel, B. MapMan Visualization of RNA-Seq Data Using Mercator4 Functional Annotations. In Solanum tuberosum: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2021; pp. 195–212. [Google Scholar]
- Griffith, M.; Walker, J.R.; Spies, N.C.; Ainscough, B.J.; Griffith, O.L. Informatics for RNA Sequencing: A Web Resource for Analysis on the Cloud. PLoS Comput. Biol. 2015, 11, e1004393. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. TopGO: Enrichment Analysis for Gene Ontology. R Package Version 2023; Bioconductor: Buffalo, NY, USA, 2023. [Google Scholar] [CrossRef]
- Wickham, H. Getting Started with Ggplot2. In ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016; pp. 11–31. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T. Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome/Linkage Group | Number of Genes with Variations | % of All Genes on a Chromosome |
|---|---|---|
| Chr2LGI | 1252 | 37% |
| Chr6LGII | 1093 | 35% |
| Chr5LGIII | 393 | 9% |
| Chr4LGIV | 232 | 6% |
| Chr3LGV | 331 | 6% |
| Chr1LGVI | 193 | 3% |
| Chr7LGVII | 509 | 6% |
| P. sativum Gene ID | M. truncatula Homologous Gene ID | Annotation | Biological Process |
|---|---|---|---|
| evm.TU.contig_464.240 | MTR_6g034940 | Abscisic acid 8-hydroxylase 3 | ABA catabolism |
| evm.TU.scaffold_2084.189 | MTR_7g102460 | UDP-glycosyltransferase 73C1 | cytokinin catabolism |
| evm.TU.scaffold_2084.193 | MTR_7g102460 | UDP-glycosyltransferase 73C1 | cytokinin catabolism |
| evm.TU.scaffold_4814.82 | MTR_5g082520 | steroid 3-dehydrogenase (CPD) | BR biosynthesis |
| P. sativum Gene ID | M. truncatula Homologous Gene ID | Encoded Enzyme | Biological Process |
|---|---|---|---|
| evm.TU.contig_1755.59 | MTR_5g033510 | tryptophan aminotransferase | auxin biosynthesis |
| evm.TU.scaffold_258.357 | MTR_1g034120 | 2-oxoglutarate-dependent dioxygenase | gibberellin biosynthesis and metabolism |
| evm.TU.contig_363.85 | MTR_2g098490 | 4-coumarate-CoA ligase-like 9 | phenylpropanoid pathway |
| evm.TU.scaffold_1663.185 | MTR_3g021440 | Isoliquiritigenin 2’-O-methyltransferase | flavonoid biosynthesis |
| evm.TU.scaffold_341.76 | MTR_0536s001 | UDP-glucose flavonoid 3-O-glucosyltransferase | flavonoid biosynthesis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zorin, E.A.; Sulima, A.S.; Zhernakov, A.I.; Kuzmina, D.O.; Rakova, V.A.; Kliukova, M.S.; Romanyuk, D.A.; Kulaeva, O.A.; Akhtemova, G.A.; Shtark, O.Y.; et al. Genomic and Transcriptomic Analysis of Pea (Pisum sativum L.) Breeding Line ‘Triumph’ with High Symbiotic Responsivity. Plants 2024, 13, 78. https://doi.org/10.3390/plants13010078
Zorin EA, Sulima AS, Zhernakov AI, Kuzmina DO, Rakova VA, Kliukova MS, Romanyuk DA, Kulaeva OA, Akhtemova GA, Shtark OY, et al. Genomic and Transcriptomic Analysis of Pea (Pisum sativum L.) Breeding Line ‘Triumph’ with High Symbiotic Responsivity. Plants. 2024; 13(1):78. https://doi.org/10.3390/plants13010078
Chicago/Turabian StyleZorin, Evgeny A., Anton S. Sulima, Aleksandr I. Zhernakov, Daria O. Kuzmina, Valeria A. Rakova, Marina S. Kliukova, Daria A. Romanyuk, Olga A. Kulaeva, Gulnar A. Akhtemova, Oksana Y. Shtark, and et al. 2024. "Genomic and Transcriptomic Analysis of Pea (Pisum sativum L.) Breeding Line ‘Triumph’ with High Symbiotic Responsivity" Plants 13, no. 1: 78. https://doi.org/10.3390/plants13010078
APA StyleZorin, E. A., Sulima, A. S., Zhernakov, A. I., Kuzmina, D. O., Rakova, V. A., Kliukova, M. S., Romanyuk, D. A., Kulaeva, O. A., Akhtemova, G. A., Shtark, O. Y., Tikhonovich, I. A., & Zhukov, V. A. (2024). Genomic and Transcriptomic Analysis of Pea (Pisum sativum L.) Breeding Line ‘Triumph’ with High Symbiotic Responsivity. Plants, 13(1), 78. https://doi.org/10.3390/plants13010078