
Pipeline to Design Inbred Lines and F1 Hybrids of Leaf Chicory (Radicchio) Using Male Sterility and Genotyping-by-Sequencing




Abstract
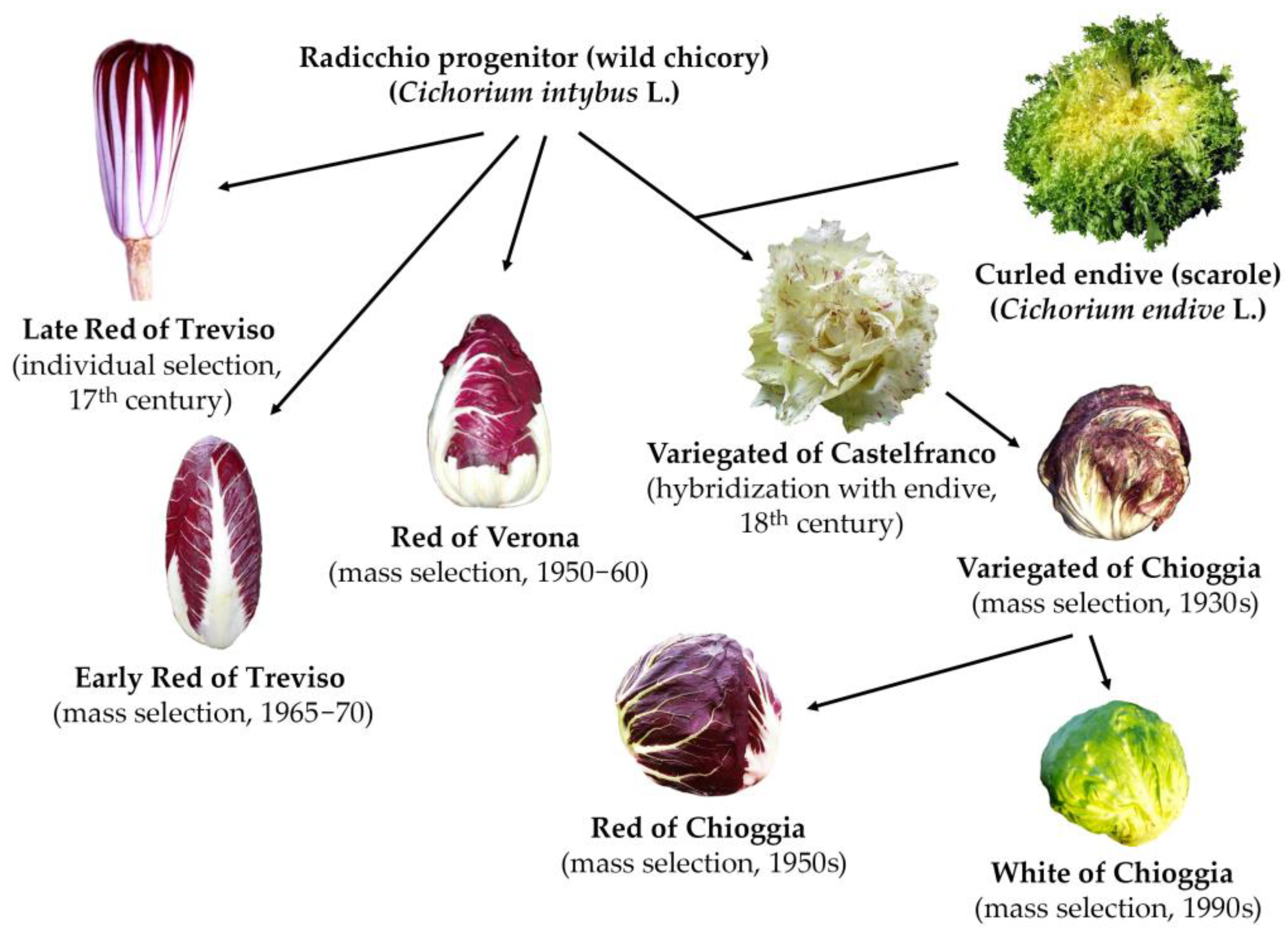
1. Introduction
2. Results
2.1. RAD Sequencing Output
2.2. Genetic Statistics and AMOVA
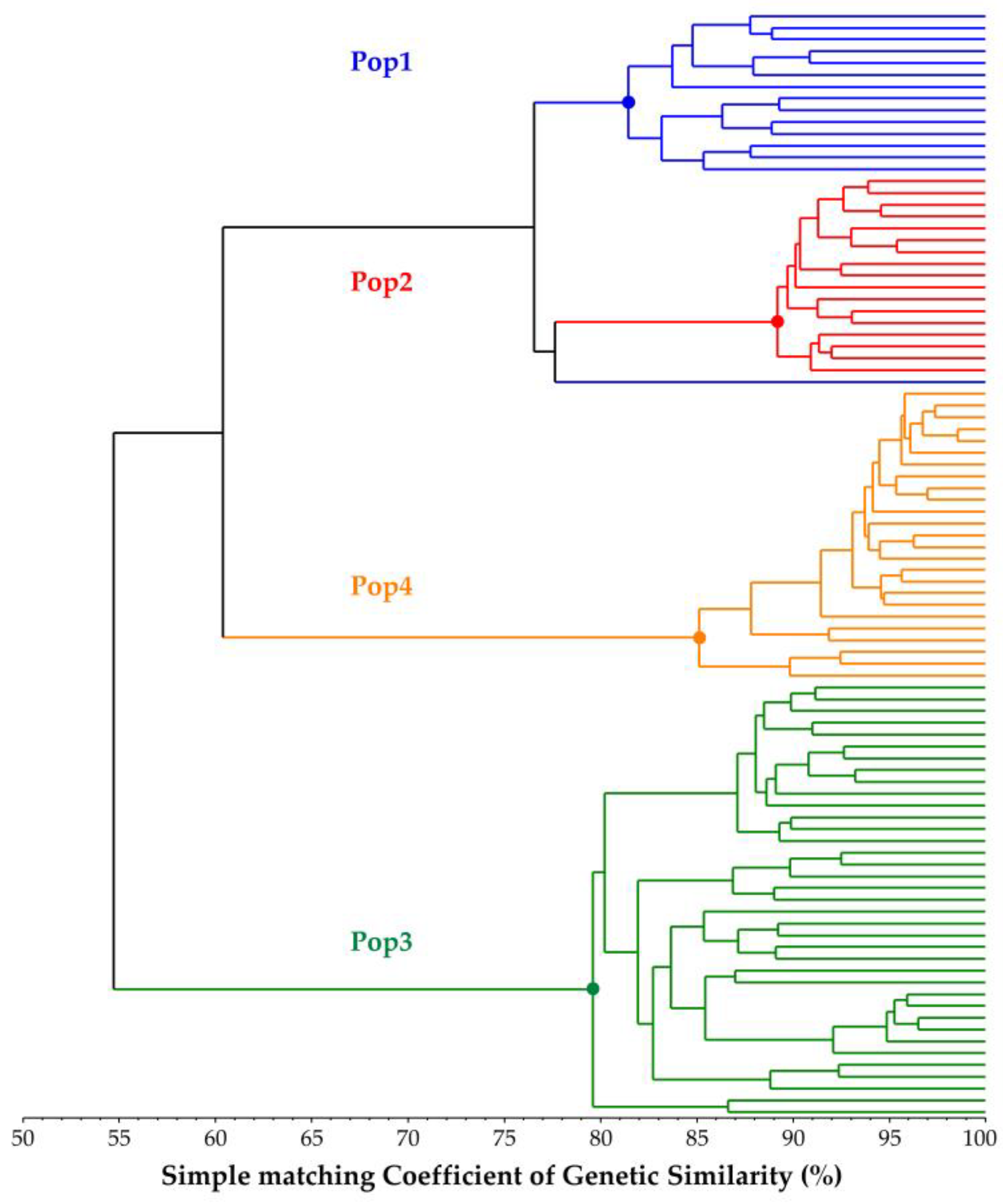
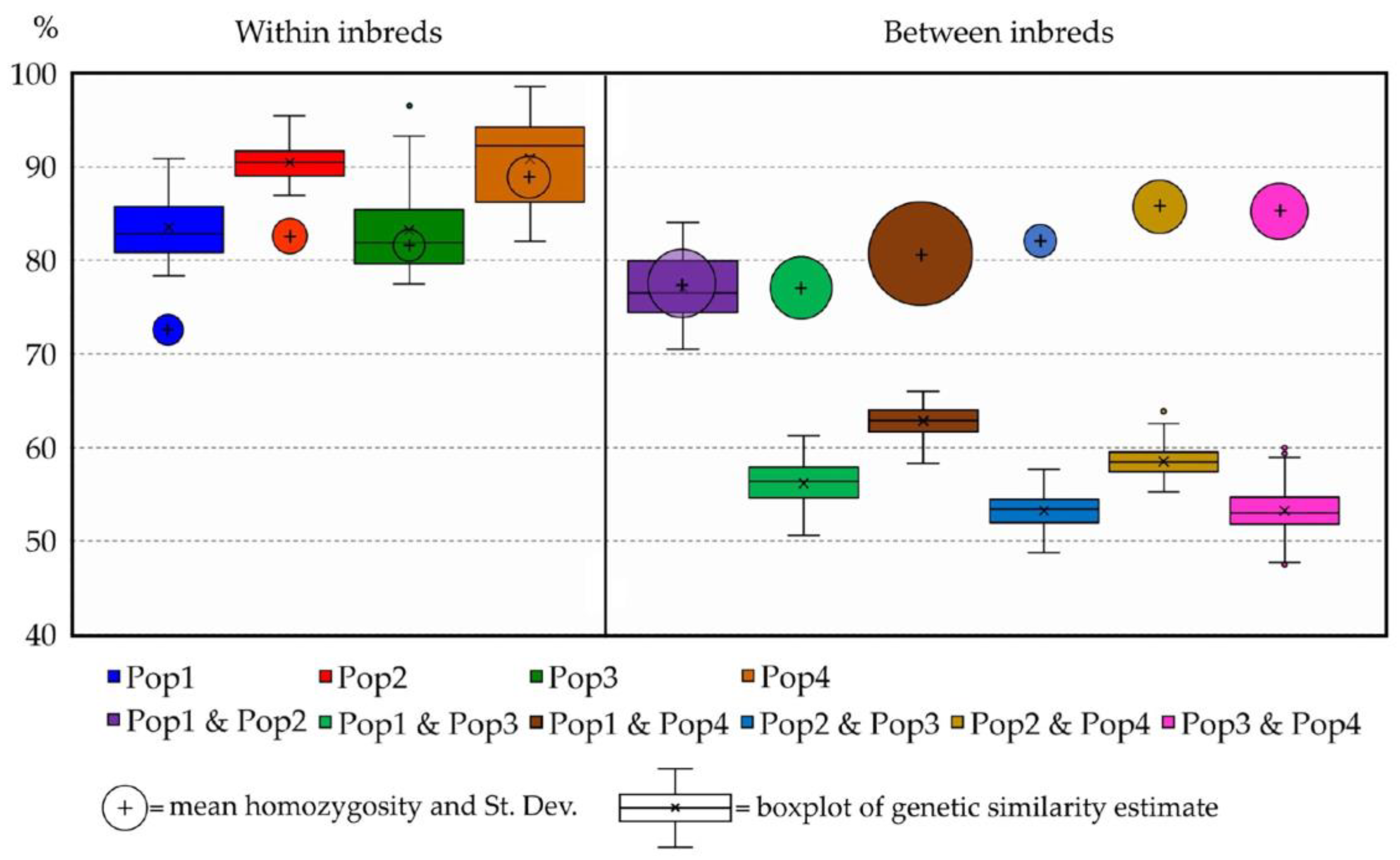
2.3. Genetic Similarity (GS) Estimates
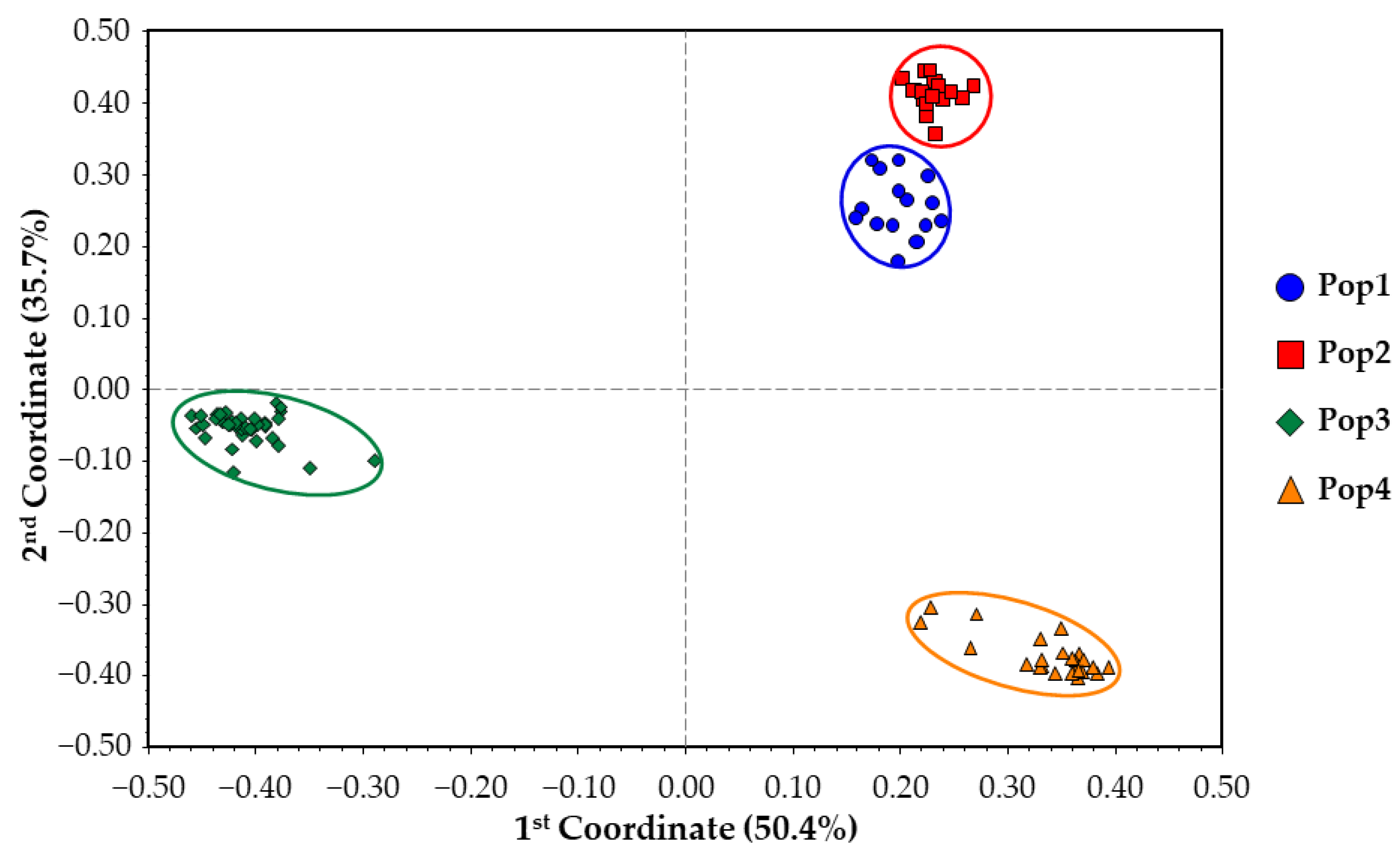
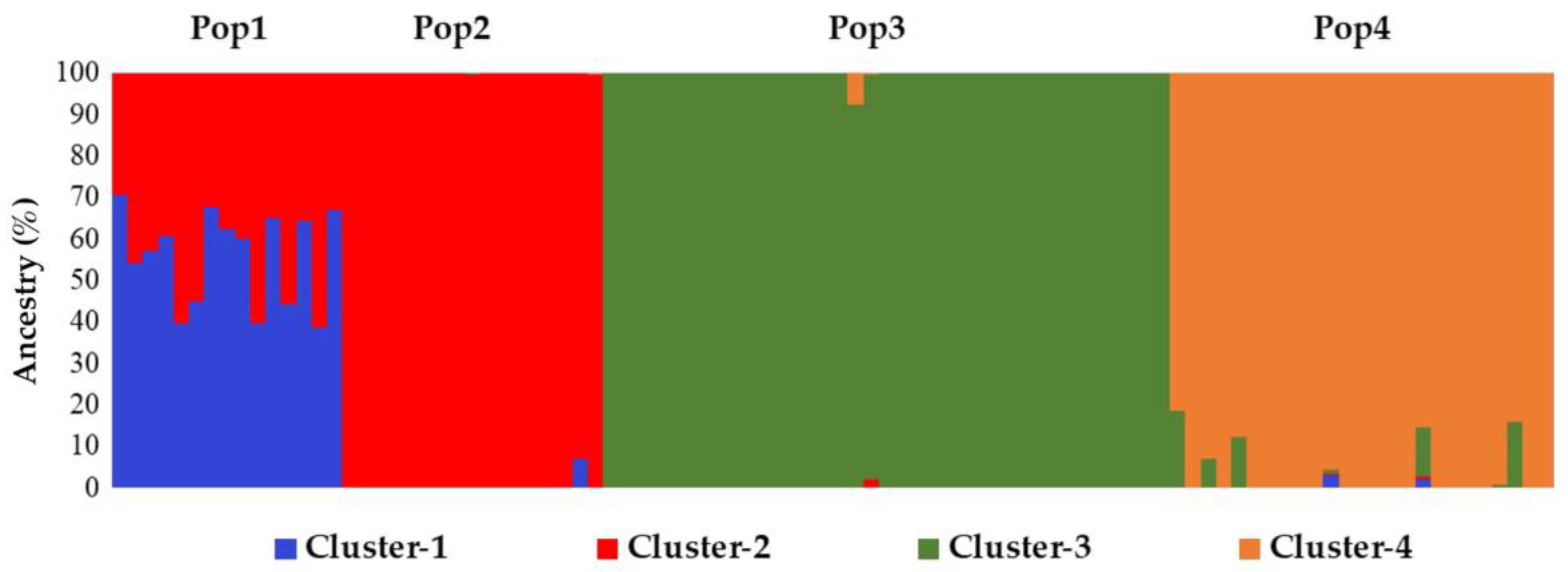
2.4. Genetic Structure Reconstruction of the Core Collection
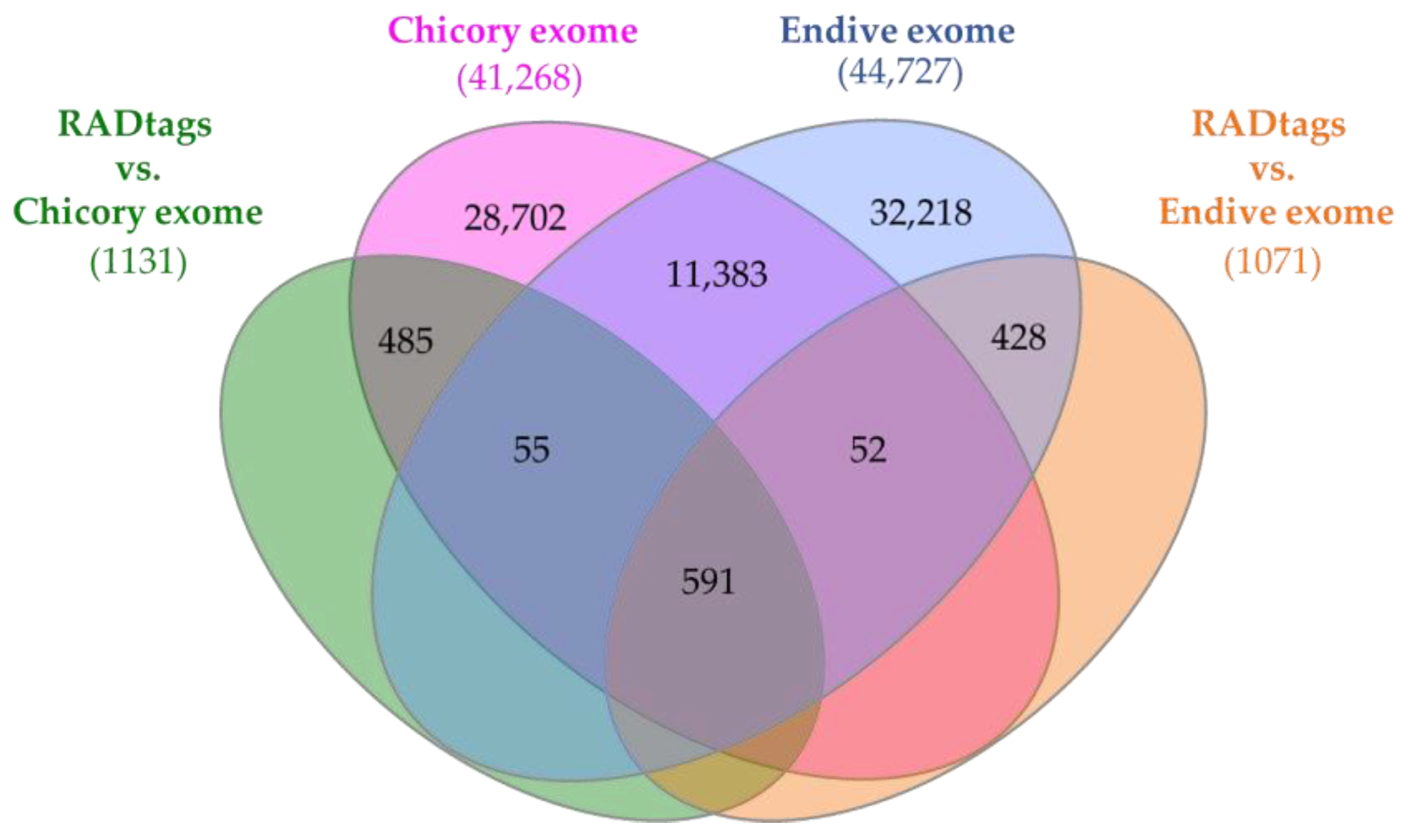
2.5. BLASTn-Based RADtag Mapping against Cichorium spp. Exomes
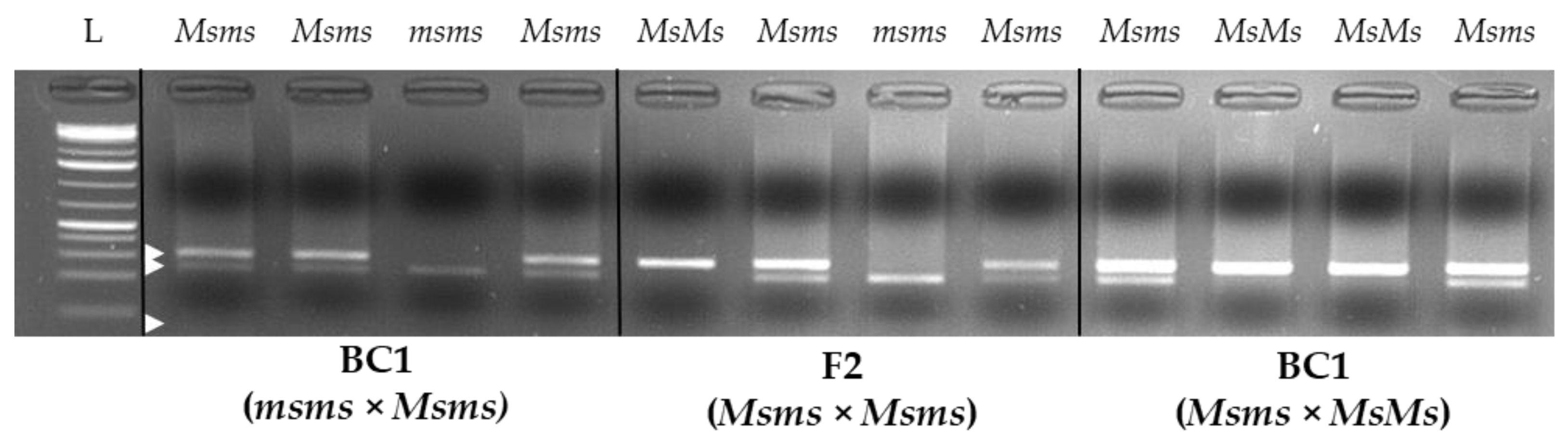
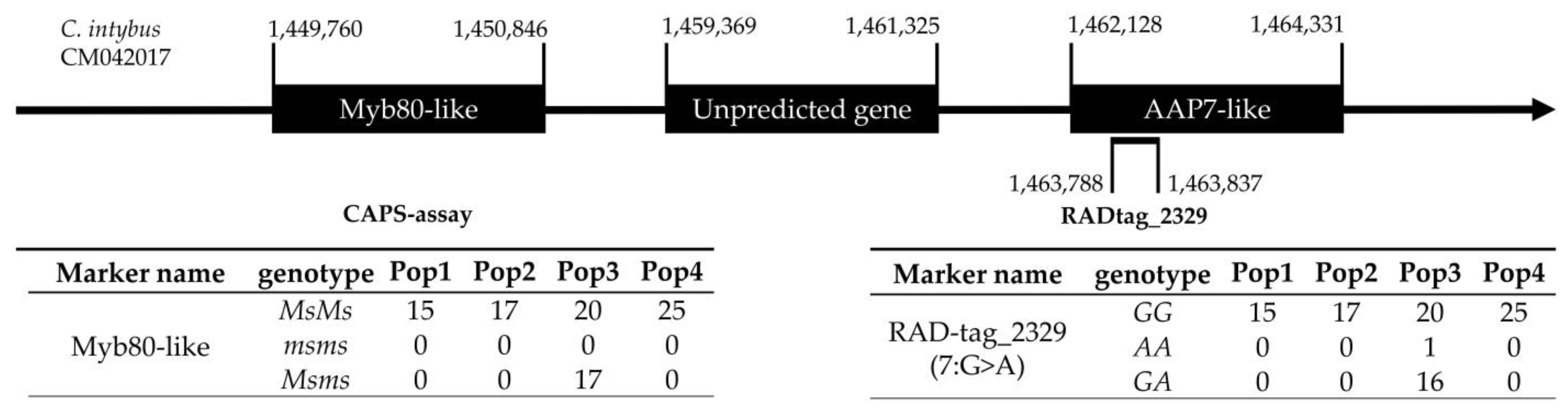
2.6. CAPS Assay and myb80-like/RADtag Association Investigation
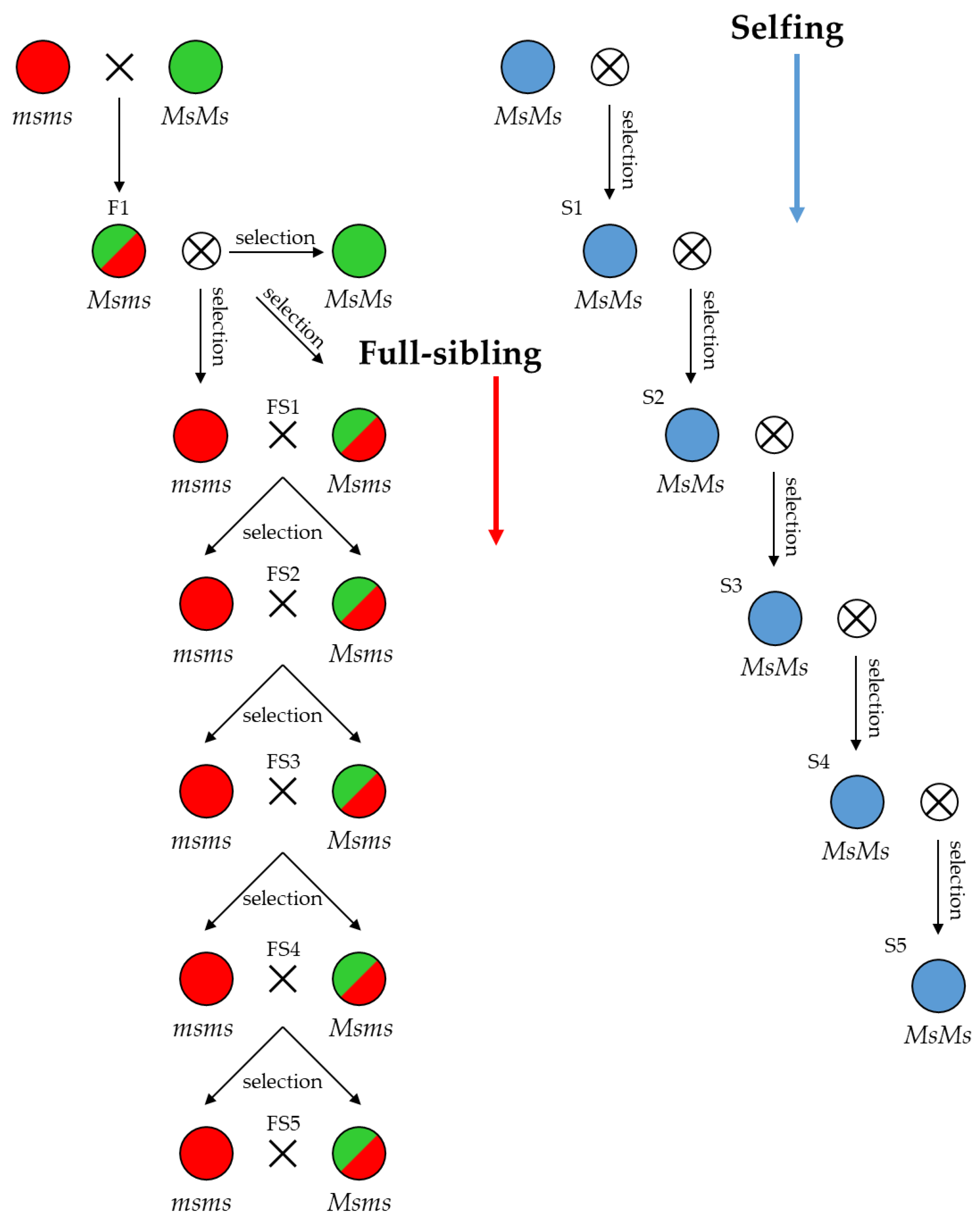
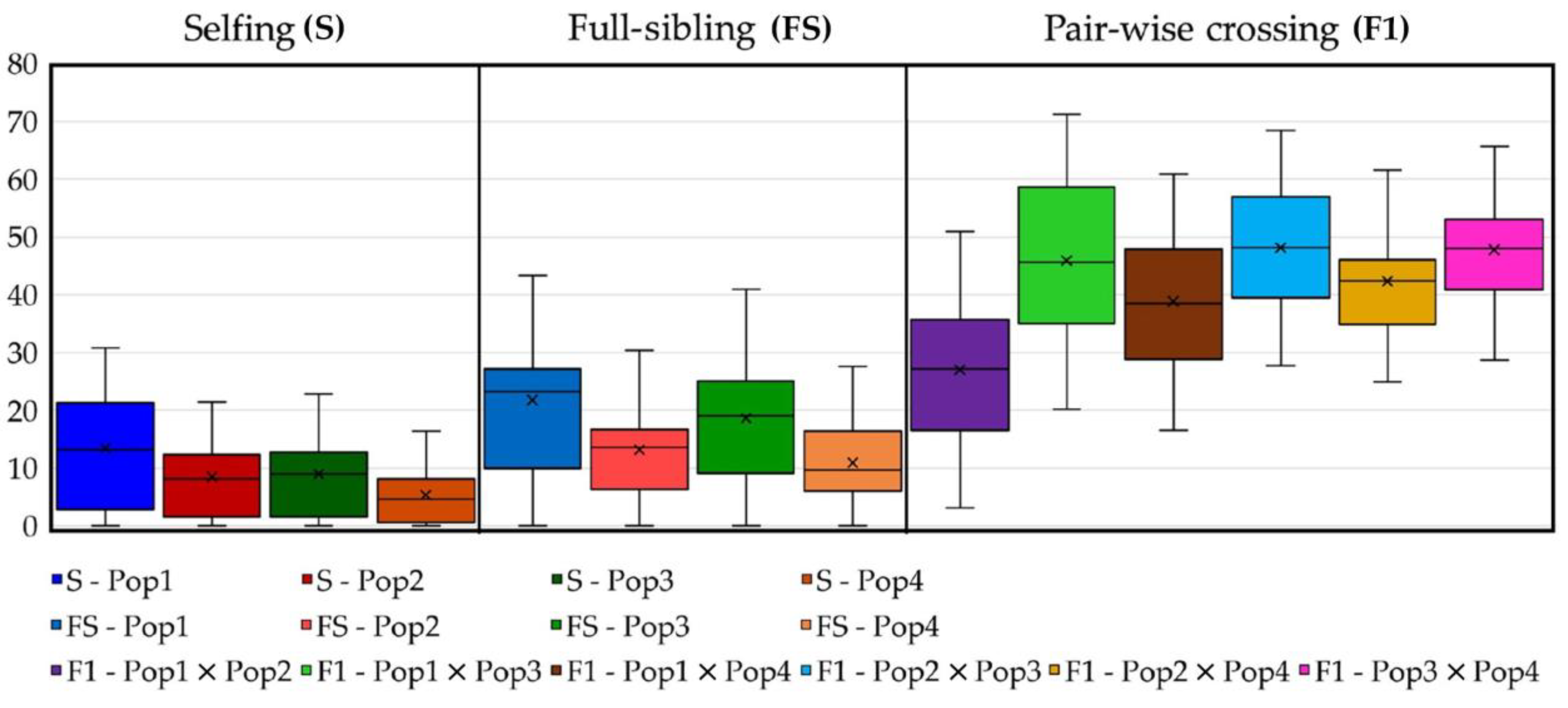
2.7. Predicting Progeny Genotypes in Planned Selfing, Full-Sibling and Pairwise Crossing Mating Strategies
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. RADseq Analysis and Data Analysis
4.3. Data Analysis and Genetic Statistics
4.4. BLASTn Analysis and RADtag Mapping
4.5. Cross-Strategy Planning for Breeding Purposes
4.6. CAPS Screening Assay for Male-Sterility Identification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bianchedi, A. I radicchi di Treviso: Storia, coltivazione, forzatura e commercio. Ital. Agric. 1961, 98, 37–51. [Google Scholar]
- Pimpini, F.; Chillemi, G.; Lazzarin, R.; Bertolini, P.; Marchetti, C. Il Radicchio Rosso di Chioggia. Aspetti Tecnici ed Economici di Produzione e Conservazione; Veneto Agricoltura: Legnaro, Padova, 2001. [Google Scholar]
- Lucchin, M.; Varotto, S.; Barcaccia, G.; Parrini, P. Chicory and Endive. In Vegetables I: Asteraceae, Brassicaceae Chenopodi-Caceae; Prohens, J., Nuez, F., Eds.; Springer: New York, NY, USA, 2008; Volume 1, pp. 3–48. [Google Scholar]
- Barcaccia, G.; Ghedina, A.; Lucchin, M. Current Advances in Genomics and Breeding of Leaf Chicory (Cichorium intybus L.). Agric.-Basel 2016, 6, 50. [Google Scholar] [CrossRef]
- Basso, A.; Scariolo, F.; Negrisolo, E.; Lucchin, M.; Barcaccia, G. Molecular Relationships and Genetic Diversity Analysis of Venetian Radicchio (Leaf Chicory, Cichorium intybus subsp. intybus var. sylvestre, 2n = 2x = 18) Biotypes. Diversity 2022, 14, 175. [Google Scholar] [CrossRef]
- Varotto, S.; Pizzoli, L.; Lucchin, M.; Parrini, P. The incompatibility system in Italian red chicory (Cichorium intybus L). Plant Breed. 1995, 114, 535–538. [Google Scholar] [CrossRef]
- Barcaccia, G.; Varotto, S.; Soattin, M.; Lucchin, M.; Parrini, P. Genetic and molecular studies of sporophytic self-incompatibility in Cichorium intybus L. In Proceedings of the Eucarpia Meeting on Leafy Vegetables Genetics and Breeding, Noordwijkerhout, The Netherlands, 19–21 March 2003. [Google Scholar]
- Barcaccia, G.; Pallottini, L.; Soattin, M.; Lazzarin, R.; Parrini, P.; Lucchin, M. Genomic DNA fingerprints as a tool for identifying cultivated types of radicchio (Cichorium intybus L.) from Veneto, Italy. Plant Breed. 2003, 122, 178–183. [Google Scholar] [CrossRef]
- Barcaccia, G.; Tiozzo Caenazzo, S. New Male Sterile Chicorium spp. Mutant, Parts or Derivatives, Where Male Sterility Is due to a Nuclear Recessive mutation Linked to a Polymorphic Genetic Marker, Useful for Producing Mutant F1 Hybrids of Chicorium spp. WO2012163389-A1, 6 December 2012. [Google Scholar]
- Barcaccia, G.; Tiozzo Caenazzo, S. New Male Sterile Mutant of Leaf Chicory, Including Radicchio, Used to Produce Chicory Plants and Seeds with traits Such as Male-Sterility Exhibiting Cytological Phenotype with Shapeless, Small and Shrunken Microspores in Dehiscent Anthers. U.S. Patent 2014157448-A1, 5 June 2014. [Google Scholar]
- Palumbo, F.; Qi, P.; Pinto, V.B.; Devos, K.M.; Barcaccia, G. Construction of the First SNP-Based Linkage Map Using Genotyping-by-Sequencing and Mapping of the Male-Sterility Gene in Leaf Chicory. Front. Plant Sci 2019, 10, 276. [Google Scholar] [CrossRef]
- Eenink, A.H. Compatibility and incompatibility in witloof-chicory (Cichorium intybus L.). 3. Gametic competition after mixed pollinations and double pollinations. Euphytica 1982, 31, 773–786. [Google Scholar] [CrossRef]
- Eenink, A.H. Compatibility and incompatibility in witloof chicory (Cichorium intybus L.). 4. Formation of self-seeds on a self-incompatible and a moderately self-compatible genotype after double and triple pollinations. Euphytica 1984, 33, 161–167. [Google Scholar] [CrossRef]
- Coppens d’Eeckenbrugge, G. The progamic phase in Cichorium intybus L. Pollen tube growth in the style, incompatibility reaction and gametophytic competition. Euphytica 1990, 48, 17–23. [Google Scholar] [CrossRef]
- Castaño, C.I.; Demeulemeester, M.A.C.; De Proft, M.P. Incompatibility reactions and genotypic identity status of five commercial chicory (Cichorium intybus L.) hybrids. Sci. Hortic. 1997, 72, 1–9. [Google Scholar] [CrossRef]
- Patella, A.; Scariolo, F.; Palumbo, F.; Barcaccia, G. Genetic Structure of Cultivated Varieties of Radicchio (Cichorium intybus L.): A Comparison between F1 Hybrids and Synthetics. Plants 2019, 8, 213. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.F. Dominance of Linked Factors as a Means of Accounting for Heterosis. Proc. Natl. Acad. Sci. USA 1917, 3, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Cadalen, T.; Morchen, M.; Blassiau, C.; Clabaut, A.; Scheer, I.; Hilbert, J.L.; Hendriks, T.; Quillet, M.C. Development of SSR markers and construction of a consensus genetic map for chicory (Cichorium intybus L.). Mol. Breed. 2010, 25, 699–722. [Google Scholar] [CrossRef]
- Gonthier, L.; Blassiau, C.; Morchen, M.; Cadalen, T.; Poiret, M.; Hendriks, T.; Quillet, M.C. High-density genetic maps for loci involved in nuclear male sterility (NMS1) and sporophytic self-incompatibility (S-locus) in chicory (Cichorium intybus L., Asteraceae). Theor. Appl. Genet. 2013, 126, 2103–2121. [Google Scholar] [CrossRef] [PubMed]
- Muys, C.; Thienpont, C.N.; Dauchot, N.; Maudoux, O.; Draye, X.; Van Cutsem, P. Integration of AFLPs, SSRs and SNPs markers into a new genetic map of industrial chicory (Cichorium intybus L. var. sativum). Plant Breed. 2014, 133, 130–137. [Google Scholar] [CrossRef]
- Galla, G.; Ghedina, A.; Tiozzo, S.C.; Barcaccia, G. Toward a First High-quality Genome Draft for Marker-assisted Breeding in Leaf Chicory, Radicchio (Cichorium intybus L.). In Plant Genomics; Abdurakhmonov, I.Y., Ed.; IntechOpen: London, UK, 2016. [Google Scholar] [CrossRef]
- Ghedina, A.; Galla, G.; Cadalen, T.; Hilbert, J.L.; Caenazzo, S.T.; Barcaccia, G. A method for genotyping elite breeding stocks of leaf chicory (Cichorium intybus L.) by assaying mapped microsatellite marker loci. BMC Res. Notes 2015, 8, 831. [Google Scholar] [CrossRef]
- Simko, I.; Jia, M.Y.; Venkatesh, J.; Kang, B.C.; Weng, Y.Q.; Barcaccia, G.; Lanteri, S.; Bhattarai, G.; Foolad, M.R. Genomics and Marker-Assisted Improvement of Vegetable Crops. Crit. Rev. Plant Sci. 2021, 40, 303–365. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.X.; Liu, H.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5, 484. [Google Scholar] [CrossRef]
- Chung, Y.S.; Choi, S.C.; Jun, T.H.; Kim, C. Genotyping-by-Sequencing: A Promising Tool for Plant Genetics Research and Breeding. Hortic. Environ. Biotechnol. 2017, 58, 425–431. [Google Scholar] [CrossRef]
- Kilian, A.; Wenzl, P.; Huttner, E.; Carling, J.; Xia, L.; Blois, H.; Caig, V.; Heller-Uszynska, K.; Jaccoud, D.; Hopper, C.; et al. Diversity Arrays Technology: A Generic Genome Profiling Technology on Open Platforms. In Data Production and Analysis in Population Genomics: Methods and Protocols; Pompanon, F., Bonin, A., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 67–89. [Google Scholar] [CrossRef]
- Kim, C.; Guo, H.; Kong, W.; Chandnani, R.; Shuang, L.S.; Paterson, A.H. Application of genotyping by sequencing technology to a variety of crop breeding programs. Plant Sci. 2016, 242, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Stevanato, P.; Broccanello, C.; Biscarini, F.; Del Corvo, M.; Sablok, G.; Panella, L.; Stella, A.; Concheri, G. High-Throughput RAD-SNP Genotyping for Characterization of Sugar Beet Genotypes. Plant Mol. Biol. Rep. 2014, 32, 691–696. [Google Scholar] [CrossRef]
- Yu, Q.; Ling, Y.; Xiong, Y.; Zhao, W.; Xiong, Y.; Dong, Z.; Yang, J.; Zhao, J.; Zhang, X.; Ma, X. RAD-seq as an effective strategy for heterogenous variety identification in plants-a case study in Italian Ryegrass (Lolium multiflorum). BMC Plant Biol. 2022, 22, 231. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Wang, S.; Wang, H.; Wang, A.; Jiang, F.; Liu, H.; Zhao, H.; Xu, D.; Zhang, Y. The genomes of chicory, endive, great burdock and yacon provide insights into Asteraceae palaeo-polyploidization history and plant inulin production. Mol. Ecol. Resour. 2022, 22, 3124–3140. [Google Scholar] [CrossRef]
- Scheben, A.; Batley, J.; Edwards, D. Genotyping-by-sequencing approaches to characterize crop genomes: Choosing the right tool for the right application. Plant Biotechnol. J. 2017, 15, 149–161. [Google Scholar] [CrossRef]
- Miller, M.R.; Dunham, J.P.; Amores, A.; Cresko, W.A.; Johnson, E.A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007, 17, 240–248. [Google Scholar] [CrossRef]
- Sica, P.; Galvao, A.; Scariolo, F.; Maucieri, C.; Nicoletto, C.; Pilon, C.; Sambo, P.; Barcaccia, G.; Borin, M.; Cabrera, M.; et al. Effects of Drought on Yield and Nutraceutical Properties of Beans (Phaseolus spp.) Traditionally Cultivated in Veneto, Italy. Horticulturae 2021, 7, 17. [Google Scholar] [CrossRef]
- Chen, L.; Fang, Y.; Li, X.; Zeng, K.; Chen, H.; Zhang, H.; Yang, H.; Cao, D.; Hao, Q.; Yuan, S.; et al. Identification of soybean drought-tolerant genotypes and loci correlated with agronomic traits contributes new candidate genes for breeding. Plant Mol. Biol. 2020, 102, 109–122. [Google Scholar] [CrossRef]
- Seki, K.; Komatsu, K.; Hiraga, M.; Tanaka, K.; Uno, Y.; Matsumura, H. Development of PCR-based marker for resistance to Fusarium wilt race 2 in lettuce (Lactuca sativa L.). Euphytica 2021, 217, 126. [Google Scholar] [CrossRef]
- Karikari, B.; Li, S.; Bhat, J.A.; Cao, Y.; Kong, J.; Yang, J.; Gai, J.; Zhao, T. Genome-Wide Detection of Major and Epistatic Effect QTLs for Seed Protein and Oil Content in Soybean Under Multiple Environments Using High-Density Bin Map. Int. J. Mol. Sci. 2019, 20, 979. [Google Scholar] [CrossRef]
- Esposito, S.; Cardi, T.; Campanelli, G.; Sestili, S.; Diez, M.J.; Soler, S.; Prohens, J.; Tripodi, P. ddRAD sequencing-based genotyping for population structure analysis in cultivated tomato provides new insights into the genomic diversity of Mediterranean ‘da serbo’ type long shelf-life germplasm. Hortic. Res. 2020, 7, 134. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Yang, T.; Zhang, F.; Yang, X.; Yang, C.; He, F.; Long, R.; Gao, T.; Jiang, Y.; Yang, Q.; et al. RAD-Seq-Based High-Density Linkage Maps Construction and Quantitative Trait Loci Mapping of Flowering Time Trait in Alfalfa (Medicago sativa L.). Front. Plant Sci. 2022, 13, 899681. [Google Scholar] [CrossRef] [PubMed]
- Zhigunov, A.V.; Ulianich, P.S.; Lebedeva, M.V.; Chang, P.L.; Nuzhdin, S.V.; Potokina, E.K. Development of F1 hybrid population and the high-density linkage map for European aspen (Populus tremula L.) using RADseq technology. BMC Plant Biol. 2017, 17, 180. [Google Scholar] [CrossRef]
- Pujol, V.; Forrest, K.L.; Zhang, P.; Rouse, M.N.; Hayden, M.J.; Huang, L.; Tabe, L.; Lagudah, E. Identification of a stem rust resistance locus effective against Ug99 on wheat chromosome 7AL using a RAD-Seq approach. Theor. Appl. Genet. 2015, 128, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Mir, R.R.; Hiremath, P.J.; Riera-Lizarazu, O.; Varshney, R.K. Evolving Molecular Marker Technologies in Plants: From RFLPs to GBS. In Diagnostics in Plant Breeding; Lübberstedt, T., Varshney, R.K., Eds.; Springer: Dordrecht, The Netherlands, 2013; pp. 229–247. [Google Scholar] [CrossRef]
- Patella, A.; Palumbo, F.; Ravi, S.; Stevanato, P.; Barcaccia, G. Genotyping by RAD Sequencing Analysis Assessed the Genetic Distinctiveness of Experimental Lines and Narrowed Down the Genomic Region Responsible for Leaf Shape in Endive (Cichorium endivia L.). Genes 2020, 11, 462. [Google Scholar] [CrossRef]
- Hsu, Y.M.; Wang, S.S.; Tseng, Y.C.; Lee, S.R.; Fang, H.; Hung, W.C.; Kuo, H.I.; Dai, H.Y. Assessment of genetic diversity and SNP marker development within peanut germplasm in Taiwan by RAD-seq. Sci. Rep. 2022, 12, 14495. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, T.; Li, H.; Wang, B.; Wu, S.; Li, S.; Wang, L.; Li, G.; Wu, X. SNP Identification from RAD-seq data in faba bean (Vicia faba L.). Plant Gene Trait 2021, 12, 1–8. [Google Scholar] [CrossRef]
- Sunde, J.; Yildirim, Y.; Tibblin, P.; Forsman, A. Comparing the Performance of Microsatellites and RADseq in Population Genetic Studies: Analysis of Data for Pike (Esox lucius) and a Synthesis of Previous Studies. Front. Genet. 2020, 11, 218. [Google Scholar] [CrossRef]
- Mcdermott, J.M.; Mcdonald, B.A. Gene Flow in Plant Pathosystems. Annu. Rev. Phytopathol. 1993, 31, 353–373. [Google Scholar] [CrossRef]
- Mitton, J.B. Gene Flow. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 192–196. [Google Scholar] [CrossRef]
- Rochette, N.C.; Catchen, J.M. Deriving genotypes from RAD-seq short-read data using Stacks. Nat. Protoc. 2017, 12, 2640–2659. [Google Scholar] [CrossRef]
- Rohlf, F.J. NTSYS-pc: Numerical Taxonomy and Multivariate Analysis System Version 2.1: Owner Manual; Exeter Publishing: Setauket, NY, USA, 2000. [Google Scholar]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed]
- Scariolo, F.; Palumbo, F.; Vannozzi, A.; Sacilotto, G.B.; Gazzola, M.; Barcaccia, G. Genotyping Analysis by RAD-Seq Reads Is Useful to Assess the Genetic Identity and Relationships of Breeding Lines in Lavender Species Aimed at Managing Plant Variety Protection. Genes 2021, 12, 1656. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef]
- Zhang, H.X.; Li, H.Y.; Li, Y.X. Identifying evolutionarily significant units for conservation of the endangered Malus sieversii using genome-wide RADseq data. Nord. J. Bot. 2018, 36, e01733. [Google Scholar] [CrossRef]
- Bao, W.Q.; Ao, D.; Wuyun, T.N.; Wang, L.; Bai, Y.E. Development of 85 SNP markers for the endangered plant species Prunus mira (Rosaceae) based on restriction site-associated DNA sequencing (RAD-seq). Conserv. Genet. Resour. 2020, 12, 525–527. [Google Scholar] [CrossRef]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Z.; Du, X.; Yang, H.; Han, F.; Han, Y.; Yuan, F.; Zhang, L.; Peng, S.; Guo, E. A high-density genetic map and QTL analysis of agronomic traits in foxtail millet [Setaria italica (L.) P. Beauv.] using RAD-seq. PLoS ONE 2017, 12, e0179717. [Google Scholar] [CrossRef] [PubMed]
- Basak, M.; Uzun, B.; Yol, E. Genetic diversity and population structure of the Mediterranean sesame core collection with use of genome-wide SNPs developed by double digest RAD-Seq. PLoS ONE 2019, 14, e0223757. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pop ID | N | na | ne | Ho (%) | He (%) | F | PL (%) |
|---|---|---|---|---|---|---|---|
| Pop1 | 15.00 | 1.56 | 1.43 | 25.96 | 23.43 | −0.10 | 56.48 |
| Pop2 | 17.00 | 1.36 | 1.27 | 16.33 | 14.02 | −0.14 | 36.23 |
| Pop3 | 37.00 | 1.54 | 1.38 | 17.17 | 20.77 | 0.17 | 54.01 |
| Pop4 | 25.00 | 1.37 | 1.21 | 12.09 | 11.93 | 0.07 | 37.45 |
| Mean | 23.50 | 1.46 | 1.32 | 17.89 | 17.54 | 0.01 | 46.05 |
| Hs | Ht | Fis | Fit | Fst | Nm | |
|---|---|---|---|---|---|---|
| Total | 0.18 | 0.36 | 0.03 | 0.48 | 0.48 | 0.63 |
| SE | 0.00 | 0.00 | 0.01 | 0.01 | 0.01 | 0.03 |
| Source | df | SS | MS | Est. Var. | % |
|---|---|---|---|---|---|
| Among Pops | 3 | 104,231 | 34,743 | 1523 | 73.10 |
| Within Pops | 90 | 50,439 | 560 | 560 | 26.90 |
| Total | 93 | 154,671 | 2083 | 100.00 |
| Obs. Homozygosity (%) | Population ID | Mean Genetic Similarity (%) | |||
|---|---|---|---|---|---|
| 74.04 ± 0.31 | Pop1 | 82.33 ± 0.04 | |||
| 83.67 ± 0.15 | Pop2 | 76.60 ± 0.01 | 90.12 ± 0.01 | ||
| 82.83 ± 0.13 | Pop3 | 56.46 ± 0.00 | 54.13 ± 0.00 | 82.50 ± 0.01 | |
| 87.91 ± 0.13 | Pop4 | 62.59 ± 0.01 | 58.46 ± 0.00 | 54.08 ± 0.00 | 91.05 ± 0.01 |
| Pop1 | Pop2 | Pop3 | Pop4 | ||
| BLASTn Results | RADtags (n) | CDS (n) | Avg. Ident (%) | Avg. Ident (n) | Avg. Length (bp) | Avg. Mis (n) | Avg. E-Value | Avg. Bs | Avg. Score | Ex. Spec. (n) | Ex. Spec. (%) | Shared (n/%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C. intybus exome | 1308 | 1131 | 98.00 | 62.22 | 63.50 | 1.26 | 9.04 × 10−13 | 109.60 | 120.55 | 224 | 15.15 | 1084 / 73.29% |
| C. endive exome | 1255 | 1071 | 97.23 | 61.91 | 63.70 | 1.74 | 4.24 × 10−13 | 107.73 | 118.43 | 171 | 11.56 |
| P1 Genotype * | P2 Genotype * | P1 × P2 Average He |
|---|---|---|
| 0 | 0 | 0 |
| 1 | 1 | 0 |
| 0 | 1 | 1 |
| 1 | 0 | 1 |
| 0/1 | 2 | 0.5 |
| 2 | 0/1 | 0.5 |
| 2 | 2 | 0.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scariolo, F.; Palumbo, F.; Farinati, S.; Barcaccia, G. Pipeline to Design Inbred Lines and F1 Hybrids of Leaf Chicory (Radicchio) Using Male Sterility and Genotyping-by-Sequencing. Plants 2023, 12, 1242. https://doi.org/10.3390/plants12061242
Scariolo F, Palumbo F, Farinati S, Barcaccia G. Pipeline to Design Inbred Lines and F1 Hybrids of Leaf Chicory (Radicchio) Using Male Sterility and Genotyping-by-Sequencing. Plants. 2023; 12(6):1242. https://doi.org/10.3390/plants12061242
Chicago/Turabian StyleScariolo, Francesco, Fabio Palumbo, Silvia Farinati, and Gianni Barcaccia. 2023. "Pipeline to Design Inbred Lines and F1 Hybrids of Leaf Chicory (Radicchio) Using Male Sterility and Genotyping-by-Sequencing" Plants 12, no. 6: 1242. https://doi.org/10.3390/plants12061242
APA StyleScariolo, F., Palumbo, F., Farinati, S., & Barcaccia, G. (2023). Pipeline to Design Inbred Lines and F1 Hybrids of Leaf Chicory (Radicchio) Using Male Sterility and Genotyping-by-Sequencing. Plants, 12(6), 1242. https://doi.org/10.3390/plants12061242