

Transcriptome Profiling and Chlorophyll Metabolic Pathway Analysis Reveal the Response of Nitraria tangutorum to Increased Nitrogen

 ,
,
Abstract
1. Introduction
2. Results
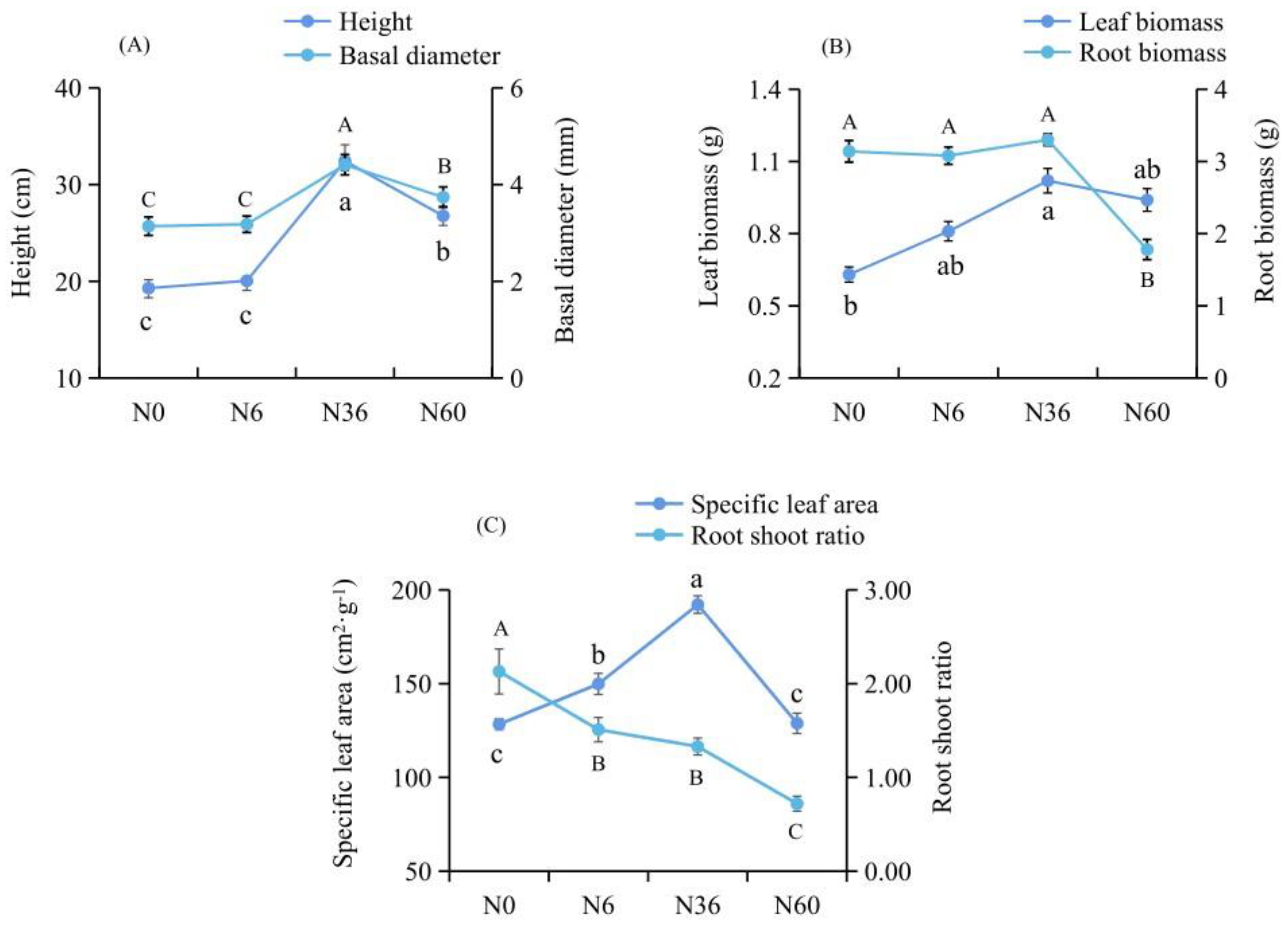
2.1. Growth and Biomass Affected by Nitrogen Treatment
2.2. RNA-Seq Analysis and Transcript Splicing
2.3. GO Functional Annotation
2.4. KOG Functional Classification
2.5. DEGs Analysis
2.6. Enrichment Analysis of DEGs in KEGG Pathways
2.7. Metabolic Responses of Porphyrin and Chlorophyll to Increased Nitrogen
2.8. Transcriptome Data Verification
3. Discussion
4. Materials and Methods
4.1. Plant Growth Conditions and Experimental Treatments
4.2. Library Preparation for Transcriptome Sequencing
4.3. Enrichment Analysis of Differentially Expressed Genes
4.4. Verification of RNA Sequencing Data by RT-qPCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gaudinier, A.; Rodriguez-Medina, J.; Zhang, L.F.; Olson, A.; Liseron-Monfils, C.; Bågman, A.M.; Foret, J.; Abbitt, S.; Tang, M.; Li, B.H.; et al. Transcriptional regulation of nitrogen-associated metabolism and growth. Nature 2018, 563, 259–264. [Google Scholar] [CrossRef]
- ContrerasLópez, O.; El Vidal, A.; Riveras, E.; Alvarez, J.M.; Moyano, T.C.; Sparks, E.E.; Medina, J.; Pasquino, A.; Benfey, P.N.; Coruzzi, G.M.; et al. Spatiotemporal analysis identifies ABF2 and ABF3 as key hubs of endodermal response to nitrate. Proc. Natl. Acad. Sci. USA 2022, 119, e2107879119. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zhou, J.Z.; Li, H.; Shi, W.; Polle, A.; Lu, M.; Sun, X.; Luo, Z. Global poplar root and leaf transcriptomes reveal links between growth and stress responses under nitrogen starvation and excess. Tree Physiol. 2015, 35, 1283–1302. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Wang, X.; Zhang, Z.; Xiong, S.; Wei, Y.; Guo, J.; Zhang, J.; Wang, L.; Ma, X.; Tegeder, M. Transcriptome and physiological studies reveal key players in wheat nitrogen use efficiency under both high and low N. J. Exp. Bot. 2021, 72, 4435–4456. [Google Scholar] [CrossRef] [PubMed]
- Niklas, K.; Owens, T.; Reich, P.B.; Cobb, E.D. Nitrogen/phosphorus leaf stoichiometry and the scaling of plant growth. Ecol. Lett. 2005, 8, 636–642. [Google Scholar] [CrossRef]
- de Bang, T.C.; Husted, S.; Laursen, K.H.; Persson, D.P.; Schjoerring, J.K. The molecular-physiological functions of mineral macronutrients and their consequences for deficiency symptoms in plants. New Phytol. 2021, 229, 2446–2469. [Google Scholar] [CrossRef]
- Cooke, J.E.K.; Martin, T.A.; Davis, J.M. Short-term physiological and developmental responses to nitrogen availability in hybrid poplar. New Phytol. 2005, 167, 41–52. [Google Scholar] [CrossRef]
- Mamashita, T.; Larocque, G.R.; DesRochers, A.; Beaulieu, J.; Thomas, B.R.; Mosseler, A.; Major, J.; Sidders, D. Short-term growth and morphological responses to nitrogen availability and plant density in hybrid poplars and willows. Biomass Bioenergy 2015, 81, 88–97. [Google Scholar] [CrossRef]
- Phoenix, G.K.; Emmett, B.A.; Britton, A.J.; Caporn, S.J.M.; Dise, N.B.; Helliwell, R.; Jones, L.; Leake, J.R.; Leith, I.D.; Sheppard, L.J.; et al. Impacts of atmospheric nitrogen deposition: Responses of multiple plant and soil parameters across contrasting ecosystems in long-term field experiments. Global Chang. Biol. 2012, 18, 1197–1215. [Google Scholar] [CrossRef]
- Shin, S.Y.; Jeong, J.S.; Lim, J.Y.; Kim, T.; Park, J.H.; Kim, J.K.; Shin, C. Transcriptomic analyses of rice (Oryza sativa) genes and non-coding RNAs under nitrogen starvation using multiple omics technologies. BMC Genom. 2018, 19, 532. [Google Scholar] [CrossRef]
- Diaz, R.J.; Rosenberg, R. Spreading dead zones and consequences for marine ecosystems. Science 2008, 321, 926–929. [Google Scholar] [CrossRef]
- Shi, X.; Cui, F.; Han, X.; He, Y.; Zhao, L.; Zhang, N.; Zhu, H.; Liu, Z.; Ma, B.; Zheng, S.; et al. Comparative genomic and transcriptomic analyses uncover the molecular basis of high nitrogen use efficiency in the wheat cultivar Kenong 9204. Mol. Plant 2022, 15, 1440–1456. [Google Scholar] [CrossRef]
- Britton, A.J.; Mitchell, R.J.; Fisher, J.M.; Riach, D.J.; Taylor, A.F. Nitrogen deposition drives loss of moss cover in alpine moss-sedge heath via lowered C:N ratio and accelerated decomposition. New Phytol. 2018, 218, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, Z.; Barberán, A.; Yang, Y.; Hu, S.; Guo, H. Nitrogen-induced acidification plays a vital role driving ecosystem functions: Insights from a 6-year nitrogen enrichment experiment in a Tibetan alpine meadow. Soil Biol. Biochem. 2021, 156, 108107. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, C.; Zheng, M.; Jiang, L.; Luo, Y. Patterns and mechanisms of responses by soil microbial communities to nitrogen addition. Soil Biol. Biochem. 2017, 115, 433–441. [Google Scholar] [CrossRef]
- Luo, L.; Ran, L.; Rasool, Q.Z.; Cohan, D.S. Integrated modeling of U.S. agricultural soil emissions of reactive nitrogen and associated impacts on air pollution, health, and climate. Environ. Sci. Technol. 2022, 56, 9265–9276. [Google Scholar] [CrossRef]
- Yang, S.; Liu, W.; Qiao, C.; Wang, J.; Deng, M.; Zhang, B.; Liu, L. The decline in plant biodiversity slows down soil carbon turnover under increasing nitrogen deposition in a temperate steppe. Funct. Ecol. 2019, 33, 1362–1372. [Google Scholar] [CrossRef]
- Dai, Z.; Su, W.; Chen, H.; Barberán, A.; Zhao, H.; Yu, M.; Yu, L.; Brookes, P.C.; Schadt, C.W.; Chang, S.X.; et al. Long-term nitrogen fertilization decreases bacterial diversity and favors the growth of Actinobacteria and Proteobacteria in agro-ecosystems across the globe. Global Chang. Biol. 2018, 24, 3452–3461. [Google Scholar] [CrossRef]
- Albinsky, D.; Kusano, M.; Higuchi, M.; Hayashi, N.; Kobayashi, M.; Fukushima, A.; Mori, M.; Ichikawa, T.; Matsuia, K.; Kurodaa, H.; et al. Metabolomic screening applied to rice FOX Arabidopsis lines leads to the identification of a gene-changing nitrogen metabolism. Mol. Plant 2010, 3, 125–142. [Google Scholar] [CrossRef]
- Chen, M.; Yin, Y.; Zhang, L.; Yang, X.; Fu, T.; Huo, X.; Wang, Y. Metabolomics and transcriptomics integration of early response of Populus tomentosa to reduced nitrogen availability. Front. Plant Sci. 2021, 12, 769748. [Google Scholar] [CrossRef]
- Luo, J.; Li, H.; Liu, T.; Polle, A.; Peng, C.; Luo, Z. Nitrogen metabolism of two contrasting poplar species during acclimation to limiting nitrogen availability. J. Exp. Bot. 2013, 64, 4207–4224. [Google Scholar] [CrossRef]
- Zhang, C.; Meng, S.; Li, M.; Zhao, Z. Transcriptomic insight into nitrogen uptake and metabolism of Populus simonii in response to drought and low nitrogen stresses. Tree Physiol. 2018, 38, 1672–1684. [Google Scholar] [CrossRef] [PubMed]
- Ruffel, S.; Krouk, G.; Ristova, D.; Shasha, D.; Birnbaum, K.D.; Coruzzi, G.M. Nitrogen economics of root foraging: Transitive closure of the nitrate–cytokinin relay and distinct systemic signaling for N supply vs. demand. Proc. Natl. Acad. Sci. USA 2011, 108, 18524–18529. [Google Scholar] [CrossRef]
- Ni, J.; Su, S.; Li, H.; Geng, Y.; Zhou, H.; Feng, Y.; Xu, X. Distinct physiological and transcriptional responses of leaves of paper mulberry (Broussonetia kazinoki × B. papyrifera) under different nitrogen supply levels. Tree Physiol. 2020, 40, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Stitt, M.; Müller, C.; Matt, P.; Gibon, Y.; Carillo, P.; Morcuende, R.; Scheible, W.; Krapp, A. Steps towards an integrated view of nitrogen metabolism. J. Exp. Bot. 2002, 53, 959–970. [Google Scholar] [CrossRef]
- Masdaux-Daubresse, C.; Daniel-Vedele, F.; Dechorgnat, J.; Chardon, F.; Gaufichon, L.; Suzuki, A. Nitrogen uptake, assimilation and remobilization in plants: Challenges for sustainable and productive agriculture. Ann. Bot. 2010, 105, 1141–1157. [Google Scholar] [CrossRef]
- Medici, A.; Marshall-Colon, A.; Ronzier, E.; Szponarski, W.; Wang, R.; Gojon, A.; Crawford, N.M.; Ruffel, S.; Coruzzi, G.M.; G Krouk, G. AtNIGT1/HRS1 integrates nitrate and phosphate signals at the Arabidopsis root tip. Nat. Commun. 2015, 6, 6274. [Google Scholar] [CrossRef]
- Xu, N.; Wang, R.; Zhao, L.; Zhang, C.; Li, Z.; Lei, Z.; Liu, F.; Guan, P.; Chu, Z.; Crawford, N.M.; et al. The Arabidopsis NRG2 protein mediates nitrate signaling and interacts with and regulates key nitrate regulators. Plant Cell. 2016, 28, 485–504. [Google Scholar] [CrossRef]
- Zhang, H.; Forde, B.G. An Arabidopsis MADS box gene that controls nutrient-induced changes in root architecture. Science 1998, 279, 407–409. [Google Scholar] [CrossRef]
- Krouk, G.; Ruffel, S.; Gutiérrez, R.A.; Gojon, A.; Crawford, N.M.; Coruzzi, G.M.; Lacombe, B. A framework integrating plant growth with hormones and nutrients. Trends Plant Sci. 2011, 16, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Qu, S.; Liang, W.; Sun, L.; Ci, D.; Ren, Z.; Fan, L.M.; and Qian, W. Nitrogen starvation induces genome-wide activation of transposable elements in Arabidopsis. J. Integr. Plant Biol. 2022, 64, 2374–2384. [Google Scholar] [CrossRef]
- Xu, G.; Fan, X.; Miller, A.J. Plant nitrogen assimilation and use efficiency. Ann. Rev. Plant Biol. 2012, 63, 153–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Garvin, D.F.; Kochian, L.V. Nitrate-induced genes in tomato roots. Array analysis reveals novel genes that may play a role in nitrogen nutrition. Plant Physiol. 2001, 127, 345–359. [Google Scholar] [CrossRef]
- Bi, Y.M.; Meyer, A.; Downs, G.S.; Shi, X.; El-Kereamy, A.; Lukens, L.; Rothstein, S.J. High throughput RNA sequencing of a hybrid maize and its parents shows different mechanisms responsive to nitrogen limitation. BMC Genom. 2014, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Li, W.; Zheng, P.; Xu, T.; Chen, L.; Liu, D.; Hussain, S.; Teng, Y. Transcriptomic analysis of ‘Suli’ pear (Pyrus pyrifolia white pear group) buds during the dormancy by RNA-Seq. BMC Genom. 2012, 13, 700. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Wu, M.; Zou, C.; Liu, X.; Shen, X.; Hayward, A.; Liu, C.; Wang, Y. Comparative analysis of anthocyanin biosynthesis during fruit development in two Lycium species. Physiol. Plant. 2014, 150, 505–516. [Google Scholar] [CrossRef]
- Wang, S.; Guan, K.; Wang, Z.; Ainsworth, E.A.; Zheng, T.; Townsend, P.A.; Li, K.; Moller, C.; Wu, G.; Jiang, C. Unique contributions of chlorophyll and nitrogen to predict crop photosynthetic capacity from leaf spectroscopy. J. Exp. Bot. 2021, 72, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wan, L.; Igathinathane, C.; Zhang, Z.; Guo, Y.; Sun, D.; Cen, H. Spatiotemporal heterogeneity of chlorophyll content and fluorescence response within rice (Oryza sativa L.) canopies under different nitrogen treatments. Front. Plant Sci. 2021, 12, 645977. [Google Scholar] [CrossRef]
- Goto, M.; Oikawa, A.; Masuda, S. Metabolic changes contributing to large biomass production in the Arabidopsis ppGpp-accumulating mutant under nitrogen deficiency. Planta 2022, 255, 48. [Google Scholar] [CrossRef]
- Zhang, C.; Huang, Y.; Xiao, Z.; Yang, H.; Hao, Q.; Yuan, S.; Chen, H.; Chen, L.; Chen, S.; Zhou, X.; et al. A GATA transcription factor from soybean (Glycine max) regulates chlorophyll biosynthesis and suppresses growth in the transgenic Arabidopsis thaliana. Plants 2020, 9, 1036. [Google Scholar] [CrossRef]
- Liu, M.; Ma, W.; Su, X.; Zhang, X.; Lu, Y.; Zhang, S.; Yan, J.; Feng, D.; Ma, L.; Taylor, A.; et al. Mutation in a chlorophyll-binding motif of Brassica ferrochelatase enhances both heme and chlorophyll biosynthesis. Cell Rep. 2022, 41, 111758. [Google Scholar] [CrossRef]
- Jia, Y.; Yu, G.; Gao, Y.; He, N.; Wang, Q.; Jiao, C.; Zuo, Y. Global inorganic nitrogen dry deposition inferred from ground-and space-based measurements. Sci. Rep. 2016, 27, 19810. [Google Scholar] [CrossRef]
- Liu, X.; Duan, L.; Mo, J.; Du, E.; Shen, J.; Lu, X.; Zhang, Y.; Zhou, X.; He, C.; Zhang, F. Nitrogen deposition and its ecological impact in China: An overview. Environ. Pollut. 2011, 159, 2251–2264. [Google Scholar] [CrossRef]
- Wang, J.; Dong, L.; Sang, W. Effects of different nitrogen regimes on competition between Ambrosia artemisiifolia, an invasive species, and two native species, Artemisia annua and Artemisia mongolica. Biodiver. Sci. 2012, 20, 3–11. (In Chinese) [Google Scholar]
- Li, Q.; Jiang, Z. Research on Plant Species of Genus Nitraria L.; China Forestry Press: Beijing, China, 2011. [Google Scholar]
- Bao, F.; Xin, Z.; Li, J.; Liu, M.; Cao, Y.; Lu, Q.; Gao, Y.; Wu, B. Effects of the simulated enhancement of precipitation on the phenology of Nitraria tangutorum under extremely dry and wet years. Plants 2021, 10, 1474. [Google Scholar] [CrossRef]
- Liu, C.; Duan, N.; Chen, X.; Li, H.; Zhao, X.; Duo, P.; Wang, J.; Li, Q. Metabolic pathways involved in the drought stress response of Nitraria tangutorum as revealed by transcriptome analysis. Forests 2022, 13, 509. [Google Scholar] [CrossRef]
- Yang, S.M.; Furukawa, I. Anatomical adaptations of three species of Chinese xerophytes (Zygophyllaceae). J. For. Res. 2006, 17, 247–251. [Google Scholar] [CrossRef]
- Yin, H.; Wang, L.; Shi, Y.; Qian, C.; Zhou, H.; Wang, W.; Ma, X.; Tran, L.S.P.; Zhang, B. The east Asian winter monsoon acts as a major selective factor in the intraspecific differentiation of drought-tolerant Nitraria tangutorum in northwest China. Plants 2020, 9, 1100. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.Q.; Wang, Y.M.; Yang, Y.L.; Zeng, Y.; Wang, Q.L.; Shao, Y.; Mei, L.J.; Shi, Y.P.; Tao, Y.D. Isolation and identification of antioxidant and α -glucosidase inhibitory compounds from fruit juice of Nitraria tangutorum. Food Chem. 2017, 227, 93–101. [Google Scholar] [CrossRef]
- Wei, Y.; Dang, X.; Wang, J.; Gao, J.; Gao, Y. Response of C:N:P in the plant-soil system and stoichiometric homeostasis of Nitraria tangutorum leaves in the oasis-desert ecotone, Northwest China. J. Arid Land 2021, 13, 934–946. [Google Scholar] [CrossRef]
- Wang, L.; Du, M.; Wang, B.; Duan, H.; Zhang, B.; Wang, D.; Li, Y.; Wang, J. Transcriptome analysis of halophyte Nitraria tangutorum reveals multiple mechanisms to enhance salt resistance. Sci. Rep. 2022, 12, 14031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.F.; Li, Q.H.; Wang, L.L.; Gao, M.Y.; Sun, G.J.; Xing, L. Effects of different nitrogen addition on bud bank traits and branching architecture of Nitraria tangutorum seedlings. For. Res. 2018, 31, 158–166. (In Chinese) [Google Scholar]
- Liu, C.G.; Sun, G.J.; Duan, N.; Li, Q.H.; Zhu, T.T.; Li, H.Q. Effects of nitrogen and phosphorus addition on chlorophyll content and photosynthetic activity of Nitraria tangutorum. Acat Agrestia Sin. 2020, 28, 694–702. (In Chinese) [Google Scholar]
- Bao, F.; Xin, Z.; Li, J.; Liu, M.; Cao, Y.; Lu, Q.; Gao, Y.; Wu, B. Preceding phenological events rather than climate drive the variations in fruiting phenology in the desert shrub Nitraria tangutorum. Plants 2022, 11, 1578. [Google Scholar] [CrossRef] [PubMed]
- Bao, F.; Xin, Z.; Liu, M.; Li, J.; Gao, Y.; Lu, Q.; Wu, B. Contrasting regulators of the onset and end of the seed release phenology of a temperate desert shrub Nitraria tangutorum. Plants 2023, 12, 88. [Google Scholar] [CrossRef]
- Bai, T.; Wang, P.; Ye, C.; Hu, S. Form of nitrogen input dominates N effects on root growth and soil aggregation: A meta-analysis. Soil Biol. Biochem. 2021, 157, 108251. [Google Scholar] [CrossRef]
- Li, H.; Yang, X.; Tang, X.; Zhang, H. Metabolomics analysis of Nitraria sibirica roots under salt stress. J. Plant Physiol. 2020, 56, 1617–1626. (In Chinese) [Google Scholar]
- Wang, W.; Li, J.; Nadeem, M.; Wang, J.; Huang, R.; Liu, Q.; Fan, W.; Zheng, H.; Yan, L.; Wang, X. The central role of GmGLP20.4 in root architecture modifications of soybean under low-nitrogen stress. Theor. Appl. Genet. 2022, 135, 4083–4093. [Google Scholar] [CrossRef]
- Gupta, A.; Rico-Medina, A.; Caño-Delgado, A.I. The physiology of plant responses to drought. Science 2020, 368, 266–269. [Google Scholar] [CrossRef]
- Kiba, T.; Krapp, A. Plant nitrogen acquisition under low availability: Regulation of uptake and root architecture. Plant Cell Physiol. 2016, 57, 707–714. [Google Scholar] [CrossRef]
- Bernard, S.M.; Habash, D.Z. The importance of cytosolic glutamine synthetase in nitrogen assimilation and recycling. New Phytol. 2009, 182, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Mo, Y.; Yang, X.; Zhang, H.; Wang, Y.; Li, H.; Wei, C.; Zhang, X. Transcriptome profiling of watermelon rootin response to short-term osmotic stress. PLoS ONE 2016, 11, e01663314. [Google Scholar]
- Wang, H.; Cao, G.; Prior, R.L. Oxygen radical absorbing capacity of anthocyanins. J. Agric. Food Chem. 1997, 45, 302–309. [Google Scholar] [CrossRef]
- Lei, B.; Chang, W.; Zhao, H.; Zhang, K.; Yu, J.; Yu, S.; Cai, K.; Zhang, J.; Lu, K. Nitrogen application and differences in leaf number retained after topping affect the tobacco (Nicotiana tabacum) transcriptome and metabolome. BMC Plant Biol. 2022, 22, 38. [Google Scholar] [CrossRef]
- Fromme, P.; Melkozernov, A.; Jordan, P.; Krauss, N. Structure and function of photosystem i: Interaction with its soluble electron carriers and external antenna systems. FEBS Lett. 2003, 555, 40–44. [Google Scholar] [CrossRef]
- Armbruster, U.; Pesaresi, P.; Pribil, M.; Hertle, A.; Leister, D. Update on chloroplast research: New tools, new topics, and new trends. Mol. Plant 2011, 4, 1–16. [Google Scholar] [CrossRef]
- Cocaliadis, M.F.; Fernández-Muñoz, R.; Pons, C.; Orzaez, D.; Granell, A. Increasing tomato fruit quality by enhancing fruit chloroplast function. a double-edged sword? J. Exp. Bot. 2014, 65, 4589–4598. [Google Scholar] [CrossRef]
- He, F.; Zhao, Q.; Huang, J.L.; Niu, M.X.; Feng, H.C.; Shi, Y.J.; Zhao, K.J.; Cui, X.L.; Wu, X.L.; Mi, J.X.; et al. External application of N alleviates toxicity of Cd on poplars via starch and sucrose metabolism. Tree Physiol. 2021, 41, 2126–2141. [Google Scholar] [CrossRef]
- Green, B.R.; Durnford, D.G. The chlorophyll-carotenoid proteins of oxygenic photosynthesis. Plant Biol. 1996, 47, 685–714. [Google Scholar] [CrossRef]
- Wei, S.; Li, X.; Lu, Z.; Zhang, H.; Ye, X.; Zhou, Y.; Li, J.; Yan, Y.; Pei, H.; Duan, F.; et al. A transcriptional regulator that boosts grain yields and shortens the growth duration of rice. Science 2022, 377, eabi8455. [Google Scholar] [CrossRef]
- Zhang, W.; Willows, R.D.; Deng, R.; Li, Z.; Li, M.; Wang, Y.; Guo, Y.; Shi, W.; Fan, Q.; Martin, S.S.; et al. Bilin-dependent regulation of chlorophyll biosynthesis by GUN4. Proc. Natl. Acad. Sci. USA 2021, 118, e2104443118. [Google Scholar] [CrossRef]
- Richter, A.; Peter, E.; Pörs, Y.; Lorenzen, S.; Grimm, B.; Czarnecki, O. Rapid dark repression of 5-aminolevulinic acid synthesis in green barley leaves. Plant Cell Physiol. 2010, 51, 670–681. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, S.; Song, S.; Xu, F.; Pan, Y.; Wang, H. Transcriptomic and proteomic analyses reveal new insight into chlorophyll synthesis and chloroplast structure of maize leaves under zinc deficiency stress. J. Prpteomics 2019, 199, 123–134. [Google Scholar] [CrossRef]
- Goslings, D.; Meskauskiene, R.; Kim, C.H.; Lee, K.P.; Nater, M.; Apel, K. Concurrent interactions of heme and FLU with Glu tRNA reductase (HEMAI), the target of metabolic feedback inhibition of tetrapyrrole biosynthesis, in dark-and light-grown Arabidopsis plants. Plant J. 2004, 40, 957–967. [Google Scholar] [CrossRef]
- Hedtke, B.; Alawady, A.; Chen, S.; Börnke, F.; Grimm, B. HEMA RNAi silencing reveals a control mechanism of ALA biosynthesis on Mg chelatase and Fe chelatase. Plant Mol. Biol. 2007, 64, 733–742. [Google Scholar] [CrossRef]
- Zhao, A.; Fang, Y.; Chen, X.; Zhao, S.; Dong, W.; Lin, Y.; Gong, W.; Liu, L. Crystal structure of Arabidopsis glutamyl-tRNAReductase in complex with its stimulator protein. Proc. Natl. Acad. Sci. USA 2014, 111, 6630–6635. [Google Scholar] [CrossRef]
- Gibson, L.C.; Willows, R.D.; Kannangara, C.G.; von Wettstein, D.; Hunter, C.N. Magnesium-protoporphyrin chelatase of Rhodobacter sphaeroides: Reconstitution of activity by combining the products of the bchH, -I, and -D genes expressed in Escherichia coli. Proc. Natl. Acad. Sci. USA 1995, 92, 1941–1944. [Google Scholar] [CrossRef] [PubMed]
- Schubert, H.L.; Raug, E.; Wilson, K.S.; Warren, M.J. Common chelatase design in branched tetrapyrrole pathways of heme and anaerobic cobalamin synthesis. Biochemistry 1999, 38, 10660–10669. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Luo, M.; Weinstein, J.D. Magnesium-chelatase from developing pea leaves: Characterization of a soluble extract from chloroplasts and resolution into three required protein fractions. Plant Physiol. 1998, 116, 605–615. [Google Scholar] [CrossRef]
- Masuda, T.; Takamiya, K.I. Novel insights into the enzymology, regulation and physiological functions of light-dependent protochlorophyllide oxidoreductase in angiosperms. Photosynth. Res. 2004, 81, 1–29. [Google Scholar] [CrossRef]
- Vezitskii, A.Y. Chlorophyll a formation in etiolated rye seedlings as dependent on the concentration of infiltrated chlorophyllide b. Russ. J. Plant Physiol. 2000, 47, 499–503. [Google Scholar]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Zhang, L.; Cheng, Z.; Hu, R.; Gao, Y.; Jin, C.; Yuan, S.; Sun, S.; Lai, M. Identification of key genes for oleoresin biosynthesis in high and low oleoresin-yielding slash pine based on transcriptome analysis. Forests 2022, 13, 1337. [Google Scholar] [CrossRef]
- Wang, T.; Hu, Y.; Chen, H.; Tan, J.; Xu, H.; Li, P.; Wu, D.; Jia, J.; Yang, Z. Transcriptome analysis of response to aluminum stress in Pinus massoniana. Forests 2022, 13, 837. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2002, 25, 402–408. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparative Group | KEGG Pathway | ID | DEG Number | p-Value |
|---|---|---|---|---|
| N0 vs. N6 | Ribosome | ko03010 | 72 | 1.35 × 10−5 |
| Anthocyanin biosynthesis | ko00942 | 7 | 0.000142 | |
| Carotenoid biosynthesis | ko00906 | 13 | 0.001063 | |
| Porphyrin and chlorophyll metabolism | ko00860 | 20 | 0.001957 | |
| Flavonoid biosynthesis | ko00941 | 10 | 0.002196 | |
| Taurine and hypotaurine metabolism | ko00430 | 7 | 0.010283 | |
| Proteasome | ko03050 | 19 | 0.011698 | |
| Amino sugar and nucleotide sugar metabolism | ko00520 | 27 | 0.012803 | |
| Photosynthesis–antenna proteins | ko00196 | 13 | 0.013312 | |
| Glutathione metabolism | ko00480 | 21 | 0.013332 | |
| Glycolysis/Gluconeogenesis | ko00010 | 37 | 0.013506 | |
| RNA degradation | ko03018 | 28 | 0.018797 | |
| Arachidonic acid metabolism | ko00590 | 8 | 0.021011 | |
| Inositol phosphate metabolism | ko00562 | 16 | 0.022433 | |
| N0 vs. N36 | Ribosome | ko03010 | 138 | 7.15 × 10−8 |
| Citrate cycle (TCA cycle) | ko00020 | 39 | 0.000176 | |
| Proteasome | ko03050 | 40 | 0.000208 | |
| Oxidative phosphorylation | ko00190 | 60 | 0.001279 | |
| Protein processing in endoplasmic reticulum | ko04141 | 95 | 0.002001 | |
| Phagosome | ko04145 | 45 | 0.002573 | |
| Spliceosome | ko03040 | 76 | 0.023464 | |
| Porphyrin and chlorophyll metabolism | ko00860 | 28 | 0.034622 | |
| Valine, leucine, and isoleucine biosynthesis | ko00290 | 10 | 0.039746 | |
| Alanine, aspartate, and glutamate metabolism | ko00250 | 32 | 0.045639 | |
| N0 vs. N60 | Starch and sucrose metabolism | ko00500 | 54 | 6.68 × 10−5 |
| Cutin, suberine, and wax biosynthesis | ko00073 | 13 | 0.000312 | |
| Limonene and pinene degradation | ko00903 | 10 | 0.002202 | |
| Pentose and glucuronate interconversions | ko00040 | 23 | 0.00236 | |
| Porphyrin and chlorophyll metabolism | ko00860 | 19 | 0.013013 | |
| Protein processing in endoplasmic reticulum | ko04141 | 52 | 0.021174 | |
| Stilbenoid, diarylheptanoid, and gingerol biosynthesis | ko00945 | 8 | 0.022509 | |
| Fatty acid degradation | ko00071 | 18 | 0.023562 | |
| Photosynthesis–antenna proteins | ko00196 | 13 | 0.029707 | |
| Zeatin biosynthesis | ko00908 | 5 | 0.030729 | |
| Anthocyanin biosynthesis | ko00942 | 4 | 0.033286 | |
| C5-branched dibasic acid metabolism | ko00660 | 4 | 0.036787 | |
| N6 vs. N36 | Carbon fixation in photosynthetic organisms | ko00710 | 94 | 1.86 × 10−6 |
| Pentose phosphate pathway | ko00030 | 66 | 4.71 × 10−6 | |
| Starch and sucrose metabolism | ko00500 | 121 | 8.61 × 10−6 | |
| Glycerolipid metabolism | ko00561 | 57 | 1.86 × 10−5 | |
| Carotenoid biosynthesis | ko00906 | 30 | 9.65 × 10−5 | |
| Anthocyanin biosynthesis | ko00942 | 10 | 0.001761583 | |
| Glycine, serine, and threonine metabolism | ko00260 | 54 | 0.003925873 | |
| Ether lipid metabolism | ko00565 | 21 | 0.006187502 | |
| Pyruvate metabolism | ko00620 | 63 | 0.006361691 | |
| Tryptophan metabolism | ko00380 | 28 | 0.009916273 | |
| Citrate cycle (TCA cycle) | ko00020 | 43 | 0.011383806 | |
| Arginine and proline metabolism | ko00330 | 39 | 0.015053622 | |
| Porphyrin and chlorophyll metabolism | ko00860 | 40 | 0.016717157 | |
| Amino sugar and nucleotide sugar metabolism | ko00520 | 64 | 0.02212956 | |
| Fructose and mannose metabolism | ko00051 | 52 | 0.026971638 | |
| Lysine degradation | ko00310 | 25 | 0.027587367 | |
| Pentose and glucuronate interconversions | ko00040 | 42 | 0.02856809 | |
| Betalain biosynthesis | ko00965 | 3 | 0.041318758 | |
| Nitrogen metabolism | ko00910 | 31 | 0.043273338 | |
| N6 vs. N60 | Stilbenoid, diarylheptanoid, and gingerol biosynthesis | ko00945 | 19 | 2.10 × 10−6 |
| Flavonoid biosynthesis | ko00941 | 18 | 2.22 × 10−5 | |
| Limonene and pinene degradation | ko00903 | 15 | 0.000202 | |
| Monoterpenoid biosynthesis | ko00902 | 10 | 0.000382 | |
| Anthocyanin biosynthesis | ko00942 | 8 | 0.000603 | |
| Arachidonic acid metabolism | ko00590 | 15 | 0.000738 | |
| Carotenoid biosynthesis | ko00906 | 17 | 0.002699 | |
| Glutathione metabolism | ko00480 | 33 | 0.005837 | |
| Cutin, suberine, and wax biosynthesis | ko00073 | 13 | 0.008832 | |
| Lysine biosynthesis | ko00300 | 10 | 0.009018 | |
| Ascorbate and aldarate metabolism | ko00053 | 25 | 0.009493 | |
| ABC transporters | ko02010 | 14 | 0.009879 | |
| Pentose and glucuronate interconversions | ko00040 | 28 | 0.012063 | |
| Phenylpropanoid biosynthesis | ko00940 | 32 | 0.017049 | |
| Starch and sucrose metabolism | ko00500 | 59 | 0.027008 | |
| Fatty acid degradation | ko00071 | 24 | 0.03139 | |
| Monobactam biosynthesis | ko00261 | 5 | 0.041373 | |
| Taurine and hypotaurine metabolism | ko00430 | 8 | 0.044949 | |
| N36 vs. N60 | Proteasome | ko03050 | 27 | 3.52 × 10−5 |
| Biosynthesis of unsaturated fatty acids | ko01040 | 13 | 0.001866 | |
| Steroid biosynthesis | ko00100 | 15 | 0.005451 | |
| Fatty acid elongation | ko00062 | 10 | 0.006054 | |
| Ribosome | ko03010 | 60 | 0.01207 | |
| Citrate cycle (TCA cycle) | ko00020 | 18 | 0.022708 | |
| Oxidative phosphorylation | ko00190 | 30 | 0.025972 | |
| Limonene and pinene degradation | ko00903 | 7 | 0.036772 | |
| Sesquiterpenoid and triterpenoid biosynthesis | ko00909 | 8 | 0.038735 |
| Soil Index | Mean | Soil Index | Mean |
|---|---|---|---|
| pH | 6.15 ± 0.146 | Organic carbon content (g·kg−1) | 6.20 ± 0.200 |
| Porosity (%) | 18.42 ± 0.055 | Total N content (g·kg−1) | 2.08 ± 0.035 |
| Water content (g·cm−3) | 1.03 ± 0.035 | Total P content (g·kg−1) | 0.12 ± 0.036 |
| Bulk density (%) | 49.99 ± 0.335 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Duan, N.; Chen, X.; Li, X.; Zhao, N.; Cao, W.; Li, H.; Liu, B.; Tan, F.; Zhao, X.; et al. Transcriptome Profiling and Chlorophyll Metabolic Pathway Analysis Reveal the Response of Nitraria tangutorum to Increased Nitrogen. Plants 2023, 12, 895. https://doi.org/10.3390/plants12040895
Liu C, Duan N, Chen X, Li X, Zhao N, Cao W, Li H, Liu B, Tan F, Zhao X, et al. Transcriptome Profiling and Chlorophyll Metabolic Pathway Analysis Reveal the Response of Nitraria tangutorum to Increased Nitrogen. Plants. 2023; 12(4):895. https://doi.org/10.3390/plants12040895
Chicago/Turabian StyleLiu, Chenggong, Na Duan, Xiaona Chen, Xu Li, Naqi Zhao, Wenxu Cao, Huiqing Li, Bo Liu, Fengsen Tan, Xiulian Zhao, and et al. 2023. "Transcriptome Profiling and Chlorophyll Metabolic Pathway Analysis Reveal the Response of Nitraria tangutorum to Increased Nitrogen" Plants 12, no. 4: 895. https://doi.org/10.3390/plants12040895
APA StyleLiu, C., Duan, N., Chen, X., Li, X., Zhao, N., Cao, W., Li, H., Liu, B., Tan, F., Zhao, X., & Li, Q. (2023). Transcriptome Profiling and Chlorophyll Metabolic Pathway Analysis Reveal the Response of Nitraria tangutorum to Increased Nitrogen. Plants, 12(4), 895. https://doi.org/10.3390/plants12040895