

Applicability of Different Methods for Quantifying Virucidal Efficacy Using MENNO Florades and Tomato Brown Rugose Fruit Virus as an Example

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results
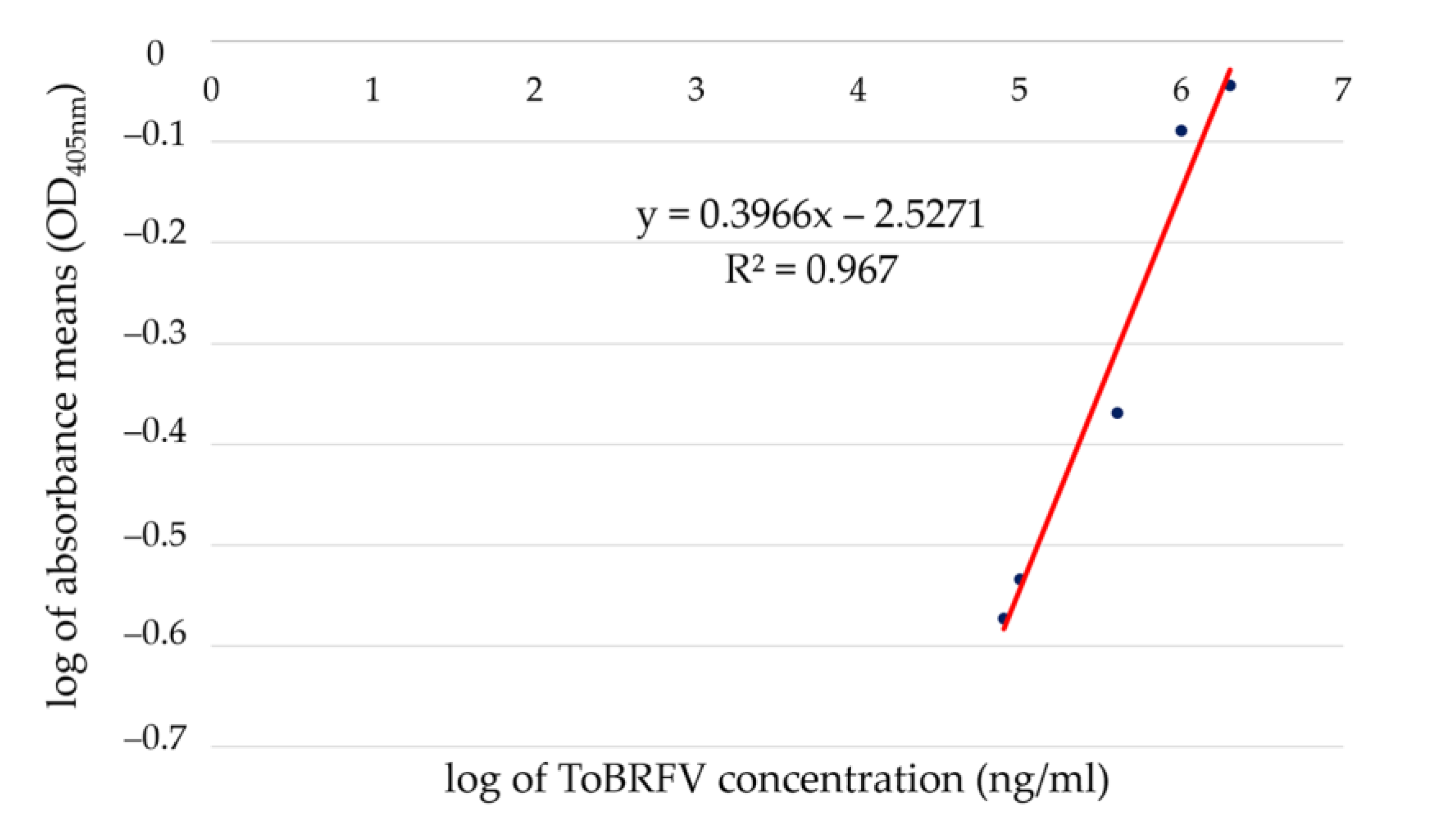
2.1. Quantitative Double Antibody Sandwitch—ELISA (qELISA)
2.2. Quantification of ToBRFV Concentration via Infectivity (Local Lesion Assay)
2.3. RNA Extraction
2.4. End Point RT-PCR, Cloning, and Sequencing
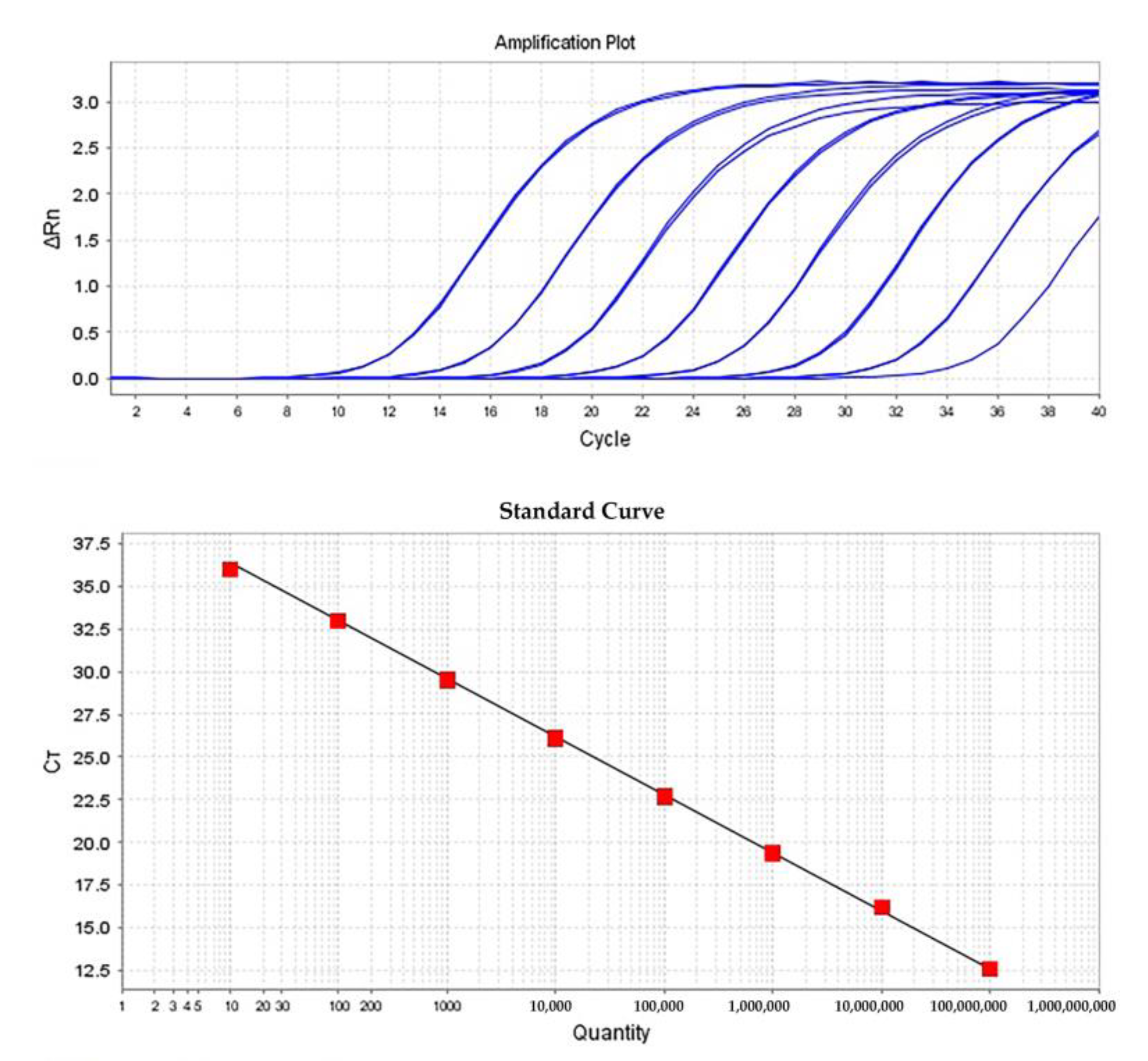
2.5. Optimization of RT-qPCR
2.6. Quantification of ToBRFV Genomic RNA by RT-qPCR
3. Discussion
4. Materials and Methods
4.1. Virus Source
4.2. Quantitative ELISA
4.3. Local Lesion Tests and Models Used for Quantifications Based on the Bioassay
4.4. Disinfection Test
4.5. RNA Isolation and cDNA Synthesis
4.6. Primers and Probe Design
4.7. Cloning and Sequencing of the ToBRFV Partial Genome Used as Standard for RT-qPCR
4.8. Plasmid Standard Curve
4.9. In Vitro Transcription and RNA Standard Curve
4.10. Absolute RT-qPCR
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shishido, M. Plant disease management in protected horticulture. Hortic. Res. 2011, 65, 7–18. [Google Scholar]
- Anderson, P.K.; Cunningham, A.A.; Patel, N.G.; Morales, F.J.; Epstein, P.R.; Daszak, P. Emerging infectious diseases of plants: Pathogen pollution, climate change and agrotechnology drivers. Trends Ecol. Evol. 2004, 19, 535–544. [Google Scholar] [CrossRef]
- FAO. Agricultural Production Statistics 2000–2020; FAO: Rome, Italy, 2022. [Google Scholar]
- Salem, N.; Mansour, A.; Ciuffo, M.; Falk, B.W.; Turina, M. A new tobamovirus infecting tomato crops in Jordan. Arch. Virol. 2016, 161, 503–506. [Google Scholar] [CrossRef]
- Avni, B.; Gelbart, D.; Sufrin-Ringwald, T.; Zinger, A.; Chen, L.; MacHbash, Z.; Bekelman, I.; Segoli, M.; Dombrovsky, A.; Kamenetsky, R.; et al. Tomato genetic resistance to tobamoviruses is compromised. Acta Hortic. 2021, 1316, 89–98. [Google Scholar] [CrossRef]
- Jones, R.A.C. Global Plant Virus Disease Pandemics and Epidemics. Plants 2021, 10, 233. [Google Scholar] [CrossRef]
- EPPO. PM 7/146 (1) Tomato brown rugose fruit virus. EPPO Bull. 2021, 51, 178–197. [Google Scholar] [CrossRef]
- Panno, S.; Caruso, A.G.; Barone, S.; Lo Bosco, G.; Rangel, E.A.; Davino, S. Spread of Tomato Brown Rugose Fruit Virus in Sicily and Evaluation of the Spatiotemporal Dispersion in Experimental Conditions. Agronomy 2020, 10, 834. [Google Scholar] [CrossRef]
- Ehlers, J.; Nourinejhad Zarghani, S.; Kroschewski, B.; Büttner, C.; Bandte, M. Cleaning of Tomato brown rugose fruit virus (ToBRFV) from Contaminated Clothing of Greenhouse Employees. Horticulturae 2022, 8, 751. [Google Scholar] [CrossRef]
- Dombrovsky, A.; Smith, E. Seed transmission of Tobamoviruses: Aspects of global disease distribution. Adv. Seed Biol. 2017, 12, 233–260. [Google Scholar]
- Balique, F.; Colson, P.; Raoult, D. Tobacco mosaic virus in cigarettes and saliva of smokers. J. Clin. Virol. 2012, 55, 374–376. [Google Scholar] [CrossRef]
- Zhang, S.; Griffiths, J.S.; Marchand, G.; Bernards, M.A.; Wang, A. Tomato brown rugose fruit virus: An emerging and rapidly spreading plant RNA virus that threatens tomato production worldwide. Mol. Plant Pathol. 2022, 23, 1262–1277. [Google Scholar] [CrossRef]
- Castello, J.D.; Rogers, S.O.; Starmer, W.T.; Catranis, C.M.; Ma, L.; Bachand, G.D.; Zhao, Y.; Smith, J.E. Detection of tomato mosaic tobamovirus RNA in ancient glacial ice. Polar Biol. 1999, 22, 207–212. [Google Scholar] [CrossRef]
- Ilyas, R.; Rohde, M.J.; Richert-Poggeler, K.R.; Ziebell, H. To Be Seen or Not to Be Seen: Latent Infection by Tobamoviruses. Plants 2022, 11, 2166. [Google Scholar] [CrossRef]
- Creager, A.N.; Scholthof, K.-B.G.; Citovsky, V.; Scholthof, H.B. Tobacco mosaic virus: Pioneering research for a century. Plant Cell 1999, 11, 301–308. [Google Scholar] [CrossRef]
- Mrkvová, M.; Hančinský, R.; Grešíková, S.; Kaňuková, Š.; Barilla, J.; Glasa, M.; Hauptvogel, P.; Kraic, J.; Mihálik, D. Evaluation of new polyclonal antibody developed for serological diagnostics of tomato mosaic virus. Viruses 2022, 14, 1331. [Google Scholar] [CrossRef]
- Moriones, E.; Verdin, E. Viral Diseases. In Integrated Pest and Disease Management in Greenhouse Crops; Gullino, M.L., Albajes, R., Nicot, P.C., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 3–31. [Google Scholar]
- Lewandowski, D.J.; Hayes, A.J.; Adkins, S. Surprising results from a search for effective disinfectants for tobacco mosaic virus–contaminated tools. Plant Dis. 2010, 94, 542–550. [Google Scholar] [CrossRef]
- Kamenova, I.; Adkins, S. Transmission, in planta distribution, and management of Hibiscus latent Fort Pierce virus, a novel tobamovirus isolated from Florida hibiscus. Plant Dis. 2004, 88, 674–679. [Google Scholar] [CrossRef]
- Hu, J.; Ferreira, S.; Xu, M.; Lu, M.; Iha, M.; Pflum, E.; Wang, M. Transmission, movement and inactivation of cymbidium mosaic and odontoglossum ringspot viruses. Plant Dis. 1994, 78, 633–636. [Google Scholar] [CrossRef]
- Coutts, B.; Kehoe, M.; Jones, R. Zucchini yellow mosaic virus: Contact transmission, stability on surfaces, and inactivation with disinfectants. Plant Dis. 2013, 97, 765–771. [Google Scholar] [CrossRef]
- Matsuura, S.; Matsushita, Y.; Usugi, T.; Tsuda, S. Disinfection of Tomato chlorotic dwarf viroid by chemical and biological agents. Crop Prot. 2010, 29, 1157–1161. [Google Scholar] [CrossRef]
- Wintermantel, W. A comparison of disinfectants to prevent spread of potyviruses in greenhouse tomato production. Plant Health Prog. 2011, 12, 19. [Google Scholar] [CrossRef]
- Baysal-Gurel, F.; Kurowski, C.; Li, R.; Ling, K.-S.; Miller, S. Developing hygiene protocols against mechanically transmitted pathogens in greenhouse tomato production systems. In Proceedings of the IV International Symposium on Tomato Diseases, Orlando, FL, USA, 24–27 June 2013; pp. 275–280. [Google Scholar]
- Ling, K.S. Recent emergence of seed-borne viruses and viroids on tomato, seed health tests and their implications in global seed trade. Acta Hortic. 2021, 1316, 127–133. [Google Scholar] [CrossRef]
- Samarah, N.; Sulaiman, A.; Salem, N.M.; Turina, M. Disinfection treatments eliminated tomato brown rugose fruit virus in tomato seeds. Eur. J. Plant Pathol. 2020, 159, 153–162. [Google Scholar] [CrossRef]
- Davino, S.; Caruso, A.G.; Bertacca, S.; Barone, S.; Panno, S. Tomato brown rugose fruit virus: Seed transmission rate and efficacy of different seed disinfection treatments. Plants 2020, 9, 1615. [Google Scholar] [CrossRef] [PubMed]
- Reingold, V.; Lachman, O.; Blaosov, E.; Dombrovsky, A. Seed disinfection treatments do not sufficiently eliminate the infectivity of Cucumber green mottle mosaic virus (CGMMV) on cucurbit seeds. Plant Pathol. 2015, 64, 245–255. [Google Scholar] [CrossRef]
- Cordoba-Selles, M.D.C.; García-Rández, A.; Alfaro-Fernández, A.; Jordá-Gutiérrez, C. Seed transmission of Pepino mosaic virus and efficacy of tomato seed disinfection treatments. Plant Dis. 2007, 91, 1250–1254. [Google Scholar] [CrossRef]
- Smith, E.; Luria, N.; Reingold, V.; Frenkel, O.; Koren, A.; Klein, E.; Bekelman, H.; Lachman, O.; Dombrovsky, A. Aspects in tobamovirus management in modern agriculture: Cucumber green mottle mosaic virus. In Proceedings of the XXX International Horticultural Congress IHC2018: International Symposium on Tropical and Subtropical Vegetable Production, Istanbul, Turkey, 12–16 August 2018; pp. 1–8. [Google Scholar]
- Dombrovsky, A.; Mor, N.; Gantz, S.; Lachman, O.; Smith, E. Disinfection Efficacy of Tobamovirus-Contaminated Soil in Greenhouse-Grown Crops. Horticulturae 2022, 8, 563. [Google Scholar] [CrossRef]
- Ehlers, J.; Nourinejhad Zarghani, S.; Kroschewski, B.; Büttner, C.; Bandte, M. Decontamination of Tomato Brown Rugose Fruit Virus-Contaminated Shoe Soles under Practical Conditions. Horticulturae 2022, 8, 1210. [Google Scholar] [CrossRef]
- Bandte, M.; Rodriguez, M.H.; Schuch, I.; Schmidt, U.; Buettner, C. Plant viruses in irrigation water: Reduced dispersal of viruses using sensor-based disinfection. Irrig. Sci. 2016, 34, 221–229. [Google Scholar] [CrossRef]
- Rodriguez, M.H.; Schmidt, U.; Büttner, C.; Bandte, M. Electrolytic Disinfection of Irrigation Water for Intensive Crop Production in Greenhouses as Demonstrated on Tomatoes (Solanum lycopersicum Mill). Horticulturae 2022, 8, 414. [Google Scholar] [CrossRef]
- Eicher-Sodo, M. Hydrogen Peroxide: A Grower’s Best Friend? Master’s Thesis, Wilfrid Laurier University, Waterloo, ON, Canada, 2020. [Google Scholar]
- Ling, K.-S.; Gilliard, A.C.; Zia, B. Disinfectants Useful to Manage the Emerging Tomato Brown Rugose Fruit Virus in Greenhouse Tomato Production. Horticulturae 2022, 8, 1193. [Google Scholar] [CrossRef]
- Li, R.; Baysal-Gurel, F.; Abdo, Z.; Miller, S.A.; Ling, K.-S. Evaluation of disinfectants to prevent mechanical transmission of viruses and a viroid in greenhouse tomato production. Virol. J. 2015, 12, 5. [Google Scholar] [CrossRef]
- Chanda, B.; Shamimuzzaman, M.; Gilliard, A.; Ling, K.S. Effectiveness of disinfectants against the spread of tobamoviruses: Tomato brown rugose fruit virus and Cucumber green mottle mosaic virus. Virol. J. 2021, 18, 7. [Google Scholar] [CrossRef]
- Cayanan, D.F.; Zhang, P.; Liu, W.; Dixon, M.; Zheng, Y. Efficacy of chlorine in controlling five common plant pathogens. HortScience 2009, 44, 157–163. [Google Scholar] [CrossRef]
- Motola, G.; Hafez, H.M.; Brüggemann-Schwarze, S. Efficacy of six disinfection methods against extended-spectrum beta-lactamase (ESBL) producing E. coli on eggshells in vitro. PLoS ONE 2020, 15, e0238860. [Google Scholar] [CrossRef]
- Rubio, L.; Galipienso, L.; Ferriol, I. Detection of plant viruses and disease management: Relevance of genetic diversity and evolution. Front. Plant Sci. 2020, 11, 1092. [Google Scholar] [CrossRef]
- Jeong, J.-J.; Ju, H.-J.; Noh, J. A review of detection methods for the plant viruses. Res. Plant Dis. 2014, 20, 173–181. [Google Scholar] [CrossRef]
- Clark, M.F.; Adams, A. Characteristics of the microplate method of enzyme-linked immunosorbent assay for the detection of plant viruses. J. Gen. Virol. 1977, 34, 475–483. [Google Scholar] [CrossRef]
- Copeland, R. Assaying levels of plant virus by ELISA. In Plant Virology Protocols; Springer: Heidelberg, Germany, 1998; pp. 455–460. [Google Scholar]
- Hou, Y.; Chen, S.; Zheng, Y.; Zheng, X.; Lin, J.-M. Droplet-based digital PCR (ddPCR) and its applications. TrAC Trends Anal. Chem. 2023, 158, 116897. [Google Scholar] [CrossRef]
- Vargas-Hernandez, B.Y.; Ramirez-Pool, J.A.; Nunez-Munoz, L.A.; Calderon-Perez, B.; De La Torre-Almaraz, R.; Hinojosa-Moya, J.; Xoconostle-Cazares, B.; Ruiz-Medrano, R. Development of a droplet digital polymerase chain reaction (ddPCR) assay for the detection of Tomato brown rugose fruit virus (ToBRFV) in tomato and pepper seeds. J. Virol. Methods 2022, 302, 114466. [Google Scholar] [CrossRef]
- Holmes, F.O. Local lesions in tobacco mosaic. Bot. Gaz. 1929, 87, 39–55. [Google Scholar] [CrossRef]
- Bald, J.G. The use of numbers of infections for comparing the concentration of plant virus suspensions: Dilution experiments with purified suspensions. Ann. Appl. Biol. 1937, 24, 33–55. [Google Scholar] [CrossRef]
- Gokhale, D.; Bald, J. Relationship between plant virus concentration and infectivity: A ‘growth curve’ model. J. Virol. Methods 1987, 18, 225–232. [Google Scholar] [CrossRef]
- Bald, J.; Iltis, R.; Schneider, S.; Gokhale, D.; Desjardins, P. Association of logistic and Poisson models of infection with some physical characteristics of a single component plant virus. J. Virol. Methods 1990, 27, 11–28. [Google Scholar] [CrossRef]
- Kleczkowski, A. Interpreting relationships between the concentrations of plant viruses and numbers of local lesions. Microbiology 1950, 4, 53–69. [Google Scholar] [CrossRef]
- Furumoto, W.A.; Mickey, R. A mathematical model for the infectivity-dilution curve of tobacco mosaic virus: Theoretical considerations. Virology 1967, 32, 216–223. [Google Scholar] [CrossRef]
- Furumoto, W.A.; Mickey, R. A mathematical model for the infectivity-dilution curve of tobacco mosaic virus: Experimental tests. Virology 1967, 32, 224–233. [Google Scholar] [CrossRef]
- Nourinejhad Zarghani, S.; Monavari, M.; Ehlers, J.; Hamacher, J.; Büttner, C.; Bandte, M. Comparison of Models for Quantification of Tomato Brown Rugose Fruit Virus Based on a Bioassay Using a Local Lesion Host. Plants 2022, 11, 3443. [Google Scholar] [CrossRef]
- Wang, Y.M.; Ostendorf, B.; Gautam, D.; Habili, N.; Pagay, V. Plant Viral Disease Detection: From Molecular Diagnosis to Optical Sensing Technology—A Multidisciplinary Review. Remote Sens. 2022, 14, 1542. [Google Scholar]
- Kleczkowski, A. The statistical analysis of plant virus assays: A transformation to include lesion numbers with small means. Microbiology 1955, 13, 91–98. [Google Scholar] [CrossRef][Green Version]
- Luria, N.; Smith, E.; Reingold, V.; Bekelman, I.; Lapidot, M.; Levin, I.; Elad, N.; Tam, Y.; Sela, N.; Abu-Ras, A.; et al. A New Israeli Tobamovirus Isolate Infects Tomato Plants Harboring Tm-22 Resistance Genes. PLoS ONE 2017, 12, e0170429. [Google Scholar] [CrossRef] [PubMed]
- Caruso, A.G.; Bertacca, S.; Parrella, G.; Rizzo, R.; Davino, S.; Panno, S. Tomato brown rugose fruit virus: A pathogen that is changing the tomato production worldwide. Ann. Appl. Biol. 2022, 181, 258–274. [Google Scholar] [CrossRef]
- Salem, N.M.; Abumuslem, M.; Turina, M.; Samarah, N.; Sulaiman, A.; Abu-Irmaileh, B.; Ata, Y. New Weed Hosts for Tomato Brown Rugose Fruit Virus in Wild Mediterranean Vegetation. Plants 2022, 11, 2287. [Google Scholar] [CrossRef] [PubMed]
- Oladokun, J.O.; Halabi, M.H.; Barua, P.; Nath, P.D. Tomato brown rugose fruit disease: Current distribution, knowledge and future prospects. Plant Pathol. 2019, 68, 1579–1586. [Google Scholar] [CrossRef]
- Mink, G. Identification of rugose mosaic-diseased cherry trees by enzyme-linked immunosorbent assay. Plant Dis. 1980, 64, 691–694. [Google Scholar] [CrossRef]
- Conci, V.C.; Canavelli, A.E.; Balzarini, M.G. The distribution of garlic viruses in leaves and bulbs during the first year of infection. J. Phytopathol. 2010, 158, 186–193. [Google Scholar] [CrossRef]
- Cafrune, E.E.; Balzarini, M.; Conci, V.C. Changes in the concentration of an Allexivirus during the crop cycle of two garlic cultivars. Plant Dis. 2006, 90, 1293–1296. [Google Scholar] [CrossRef]
- Lomonossoff, G.P.; Wege, C. TMV Particles: The Journey from Fundamental Studies to Bionanotechnology Applications. In Advances in Virus Research; Palukaitis, P., Roossinck, M.J., Eds.; Academic Press: New York, NY, USA, 2018; Volume 102, pp. 149–176. [Google Scholar]
- Clark, M.F.; Barbara, D.J. A method for the quantitative analysis of ELISA data. J. Virol. Methods 1987, 15, 213–222. [Google Scholar] [CrossRef]
- Ferriol, I.; Ruiz-Ruiz, S.; Rubio, L. Detection and absolute quantitation of Broad bean wilt virus 1 (BBWV-1) and BBWV-2 by real time RT-PCR. J. Virol. Methods 2011, 177, 202–205. [Google Scholar] [CrossRef]
- Bertolini, E.; Moreno, A.; Capote, N.; Olmos, A.; de Luis, A.; Vidal, E.; Pérez-Panadés, J.; Cambra, M. Quantitative detection of Citrus tristeza virus in plant tissues and single aphids by real-time RT-PCR. Eur. J. Plant Pathol. 2008, 120, 177–188. [Google Scholar] [CrossRef]
- Ruiz-Ruiz, S.; Ambrós, S.; del Carmen Vives, M.; Navarro, L.; Moreno, P.; Guerri, J. Detection and quantitation of Citrus leaf blotch virus by TaqMan real-time RT-PCR. J. Virol. Methods 2009, 160, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Vásquez, J.A.; Rubio, L.; Alfaro-Fernández, A.; Debreczeni, D.E.; Font-San-Ambrosio, I.; Falk, B.W.; Ferriol, I. Detection and absolute quantitation of Tomato torrado virus (ToTV) by real time RT-PCR. J. Virol. Methods 2015, 221, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Menzel, W.; Winter, S. Identification of novel and known tobamoviruses in tomato and other solanaceous crops using a new pair of generic primers and development of a specific RT-qPCR for ToBRFV. Acta Hortic. 2021, 1316, 143–148. [Google Scholar] [CrossRef]
- Panno, S.; Ruiz-Ruiz, S.; Caruso, A.G.; Alfaro-Fernandez, A.; Font San Ambrosio, M.I.; Davino, S. Real-time reverse transcription polymerase chain reaction development for rapid detection of Tomato brown rugose fruit virus and comparison with other techniques. PeerJ 2019, 7, e7928. [Google Scholar] [CrossRef] [PubMed]
- White, E.J.; Venter, M.; Hiten, N.F.; Burger, J.T. Modified Cetyltrimethylammonium bromide method improves robustness and versatility: The benchmark for plant RNA extraction. Biotechnol. J. 2008, 3, 1424–1428. [Google Scholar] [CrossRef]
- Nassuth, A.; Pollari, E.; Helmeczy, K.; Stewart, S.; Kofalvi, S.A. Improved RNA extraction and one-tube RT-PCR assay for simultaneous detection of control plant RNA plus several viruses in plant extracts. J. Virol. Methods 2000, 90, 37–49. [Google Scholar] [CrossRef]
- Cordeiro, M.C.R.; Silva, M.S.; de Oliveira-Filho, E.C.; de Miranda, Z.D.J.G.; de Góis Aquino, F.; da Rocha, R. Optimization of a method of total RNA extraction from Brazilian native plants rich in polyphenols and polysaccharides. In Proceedings of the Simposio Internacional Savanas Tropicais ParlaMundi, Brasilia, Brazil, 12–17 October 2008; pp. 12–17. [Google Scholar]
- Grodetskaya, T.; Fedorova, O.; Evlakov, P. Optimized method for RNA extraction from leaves of forest tree species. IOP Conf. Ser. Earth Environ. Sci. 2021, 875, 012008. [Google Scholar] [CrossRef]
- Zamboni, A.; Pierantoni, L.; De Franceschi, P. Total RNA extraction from strawberry tree (Arbutus unedo) and several other woody-plants. Iforest-Biogeosci. For. 2008, 1, 122. [Google Scholar] [CrossRef]
- Levesque-Sergerie, J.-P.; Duquette, M.; Thibault, C.; Delbecchi, L.; Bissonnette, N. Detection limits of several commercial reverse transcriptase enzymes: Impact on the low-and high-abundance transcript levels assessed by quantitative RT-PCR. BMC Mol. Biol. 2007, 8, 93. [Google Scholar] [CrossRef]
- Chipley, J.R. Sodium benzoate and benzoic acid. In Antimicrobials in Food; CRC Press: Boca Raton, FL, USA, 2020; pp. 41–88. [Google Scholar]
- Fehres, H.; Linkies, A. A mechanized two-step cleaning and disinfection process strongly minimizes pathogen contamination on wooden potato storage boxes. Crop Prot. 2018, 103, 111–114. [Google Scholar] [CrossRef]
- Celar, F.; Valic, N.; Kosmelj, K.; Gril, T. Evaluating the efficacy, corrosivity and phytotoxicity of some disinfectants against Erwinia amylovora (Burrill) Winslow et al. using a new statistical measure. J. Plant Dis. Prot. 2007, 114, 49–53. [Google Scholar] [CrossRef]
- Whelan, J.A.; Russell, N.B.; Whelan, M.A. A method for the absolute quantification of cDNA using real-time PCR. J. Immunol. Methods 2003, 278, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Caspar, D. Assembly and stability of the tobacco mosaic virus particle. Adv. Protein Chem. 1964, 18, 37–121. [Google Scholar]



{kind=link}
{kind=link}
| Sample Concentration or Name | Log of Virus Concentration | Mean Value of Reads | Log of Mean Values of Reads | Reacted | Calculated Concentration (mg/mL) | |
|---|---|---|---|---|---|---|
| Serially diluted ToBRFV particles to establish the standard curve for qELISA | 2,000,000 ng/mL | 6.3010 | 0.9045 | −0.0436 | positive * | 1.83 |
| 400,000 ng/mL | 5.6021 | 0.4274 | −0.3692 | positive | 0.47 | |
| 80,000 ng/mL | 4.9031 | 0.2674 | −0.5728 | positive | 0.085 | |
| 16,000 ng/mL | 4.2041 | 0.2456 | −0.6098 | positive | 0.017 | |
| 3200 ng/mL | 3.5052 | 0.1538 | −0.8130 | negative | NA ** | |
| 640 ng/mL | 2.8062 | 0.1031 | −0.9867 | negative | NA | |
| 128 ng/mL | 2.1072 | 0.1322 | −0.8788 | negative | NA | |
| 1,000,000 ng/mL | 6 | 0.8156 | −0.0885 | positive | 1.40 | |
| 100,000 ng/mL | 5 | 0.2922 | −0.5343 | positive | 0.11 | |
| 10,000 ng/mL | 4 | 0.1523 | −0.8173 | negative | NA | |
| Germ carriers and plant sap loaded by 1 mg ToBRFV particles and treated with 4% MENNO Florades for 16 h | metal carrier untreated with MF | 0.0962 | −1.0168 | negative | NA | |
| metal carrier disinfected with MF | 0.1045 | −0.9809 | negative | NA | ||
| plastic carrier untreated with MF | 0.1012 | −1.9208 | negative | NA | ||
| plastic carrier disinfected with MF | 0.1599 | −0.7962 | negative | NA | ||
| plant sap untreated with MF | 0.0864 | −1.0635 | negative | NA | ||
| plant sap disinfected with MF | 0.0829 | −1.0814 | negative | NA | ||
| Inoculated plants with ToBRFV | N. benthamiana plant (1:10 diluted) | 0.8069 | −0.0932 | positive | 1.37 (13.7) *** | |
| N. clevelandii plant (1:10 diluted) | 0.9091 | −0.0414 | positive | 1.85 (18.52) *** | ||
| Negative control (N. clevelandii non-inoculated) | 0.1089 | −0.9630 | negative | NA | ||
| Negative control (N. benthamiana non-inoculated) | 0.1013 | −0.9944 | negative | NA | ||
| Substrate (p-Nitrophenyl phosphate) | 0.0738 | −1.1319 | NA | NA | ||
| Virus Titer in Inoculum (mg/mL) | Calculated Virus Load in Inoculum or Remained Virus Load after Treatment with Disinfectant (mg/mL) Based on | |||
|---|---|---|---|---|
| The Kleczkowski Model [51] | The Growth Curve Model [49] | |||
| 2 | 87.130 | 2.0629 | 2.0267 | |
| Virus titer in serially diluted ToBRFV isolate PV-1236 particles used as inoculum | 0.4 | 25.909 | 0.3367 | 0.3520 |
| 0.08 | 12.477 | 0.1167 | 0.1226 | |
| 0.016 | 2.616 | 0.0130 | 0.0128 | |
| 0.0032 | 1.128 | 0.0042 | 0.0038 | |
| 0.00064 | 0.493 | 0.0014 | 0.0012 | |
| 0.000128 | 0.030 | 0.0003 | 0.0002 | |
| N. clevelandii | unknown | 85.9892 | 2.0218 | 1.9885 |
| N. benthamiana | unknown | 41.0477 | 1.7765 | 1.7647 |
| disinfected metal carrier ** | unknown | 0.00689 | 0.00001 | 0.00000 |
| disinfected plastic carrier | unknown | 0.02001 | 0.0004 | 0.00045 |
| disinfected sap | unknown | 0.09529 | 0.00019 | 0.00011 |
| Model parameters *** | N = 11,064, λ = 2, = 5.16 | N= 1134.53, β = 20, γ = 1.64 | ||
| 2.40 | 1.60 | |||
| Name | Sequence 5’ → 3’ | Start | End | Application |
|---|---|---|---|---|
| ToBRFV-1482-s | TAATCAGCAAGTTTAGTTTG | 1482 | 1501 | D, C |
| ToBRFV-1677-as | TCAGTCACTAATCTATCGTG | 1677 | 1658 | D, C |
| ToBRFV-4750-as | GGATCTTCTGAACTCTTCTA | 4750 | 4731 | C, I |
| ToBRFV-4640-s | ATACATCATGACAGAGGGTG | 4640 | 4659 | C |
| ToBRFV-6323-as | GCCTACGGATGTGTATGAAC | 6342 | 6323 | C |
| ToBRFV-KpnI-4388-s | GTTTATGGTACCAGAGAAAGAG | 4389 | 4410 | C |
| ToBRFV-HindIII-6153-as | CTCTAAGCTTACCATTGTAAACCGGATGCAC | 6173 | 6153 | C |
| ToBRFV-CP-Eco47-s | CGTAGAGTAGATGACGCAACG | 6050 | 6070 | C |
| ToBRFV-HindIII-3UTR-as | TATATAAGCTTGCATGCTGGGCCCCTACCGGGGGTTCCGGGGGAAT | 6393 | 6366 | C, I |
| ToBRFV-2703-s | AAGCCACAAGAGATAATGTTCGTA | 2703 | 2726 | Q |
| ToBRFV-2838-as | CAATTTCGCACAGAGACATAG | 2858 | 2838 | Q, I |
| ToBRFV-2760L | 6FAM-CTGACAGCGTGTTCCTTTACCG-BHQ1 | 2773 | 2752 | Q |
| Sample | Total RNA Used in cDNA (ng) | Ct1 ± (SD) 2 | Quantity ± (SD) | Calculated Virus Concentration | ||
|---|---|---|---|---|---|---|
| Copy No. per µg Total RNA | VP 3 (mg) per µg Total RNA | VP (mg) per 100 mg Tissue | ||||
| 0.1 mg virus particles (diluted 1:5000) 4 | 500 | 17.39 ± (0.087) | 2,730,724.75 ± (162233) | 2.7307 × 1010 | 0.0018 | 0.0056 |
| 0.5µg in vitro transcript RNA (diluted 1:5000) equal to 0.0126 mg particles 5 | 500 | 15.264 ± (0.077) | 6,738,004 ± (339,407.656) | 6.738 × 1010 | 0.0045 | NA 6 |
| Inoculated N. clevelandii (diluted 1:50,000) | 500 | 22.793 ± (0.272) | 45,962.41 ± (8171.118) | 4.596 × 109 | 0.0003 | 0.0023 |
| Inoculated N. benthamiana (diluted 1:5000) | 500 | 23.098 ± (0.333) | 37,752.36 ± (8331.31) | 3.775 × 109 | 0.0002 | 0.0013 |
| N. clevelandii containing 0.1 mg ToBRFV particles (diluted 1:50,000) | 1000 | 21.644 ± (0.087) | 97,568.10 ± (5498.168) | 4.88 × 109 | 0.0003 | 0.0025 |
| N. clevelandii containing 1 mg ToBRFV particles (diluted 1:50,000)(i.e., plant sap untreated with MF) | 1000 | 20.891 ± (0.068) | 160,767.54 ± (7322.144) | 8.03109 | 0.0005 | 0.0128 |
| metal carrier untreated with MF | NA | |||||
| metal carrier disinfected with MF | NA | |||||
| plastic carrier untreated with MF | NA | |||||
| plastic carrier disinfected with MF | NA | |||||
| plant sap untreated with MF | NA | |||||
| Sample Concentration or Name | qELISA mg/mL | RT-qPCR mg/mL | Bioassay Calculated Virus Concentration (mg/mL) | |
|---|---|---|---|---|
| inoculum | 1 mg/mL | 1.37 | 0.0056 † | 1 ‡ |
| Germ carriers and plant sap loaded by 1 mg ToBRFV particles and treated with 4% MENNO Florades (MF) for 16 h | metal carrier untreated with MF * | NA (negative) ** | NA (negative) *** | 0.95 |
| metal carrier disinfected with MF | NA (negative) | NA (negative) | 0.000002 | |
| plastic carrier untreated with MF | NA (negative) | NA (negative) | 0.93 | |
| plastic carrier disinfected with MF | NA (negative) | NA (negative) | 0.00011 | |
| plant sap untreated with MF | NA (negative) | NA (negative) | 1.003 | |
| plant sap disinfected with MF | NA (negative) | NA (negative) | 0.00011 | |
| Inoculated plants with ToBRFV | N. benthamiana plant | 13.7 | 0.0013 | 1.76–1.78 |
| N. clevelandii plant | 18.5 | 0.0023 | 1.99–2.02 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nourinejhad Zarghani, S.; Ehlers, J.; Monavari, M.; von Bargen, S.; Hamacher, J.; Büttner, C.; Bandte, M. Applicability of Different Methods for Quantifying Virucidal Efficacy Using MENNO Florades and Tomato Brown Rugose Fruit Virus as an Example. Plants 2023, 12, 894. https://doi.org/10.3390/plants12040894
Nourinejhad Zarghani S, Ehlers J, Monavari M, von Bargen S, Hamacher J, Büttner C, Bandte M. Applicability of Different Methods for Quantifying Virucidal Efficacy Using MENNO Florades and Tomato Brown Rugose Fruit Virus as an Example. Plants. 2023; 12(4):894. https://doi.org/10.3390/plants12040894
Chicago/Turabian StyleNourinejhad Zarghani, Shaheen, Jens Ehlers, Mehran Monavari, Susanne von Bargen, Joachim Hamacher, Carmen Büttner, and Martina Bandte. 2023. "Applicability of Different Methods for Quantifying Virucidal Efficacy Using MENNO Florades and Tomato Brown Rugose Fruit Virus as an Example" Plants 12, no. 4: 894. https://doi.org/10.3390/plants12040894
APA StyleNourinejhad Zarghani, S., Ehlers, J., Monavari, M., von Bargen, S., Hamacher, J., Büttner, C., & Bandte, M. (2023). Applicability of Different Methods for Quantifying Virucidal Efficacy Using MENNO Florades and Tomato Brown Rugose Fruit Virus as an Example. Plants, 12(4), 894. https://doi.org/10.3390/plants12040894