


Agro-Morphological and Molecular Characterization Reveal Deep Insights in Promising Genetic Diversity and Marker-Trait Associations in Fagopyrum esculentum and Fagopyrum tataricum

 ,
,  ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results
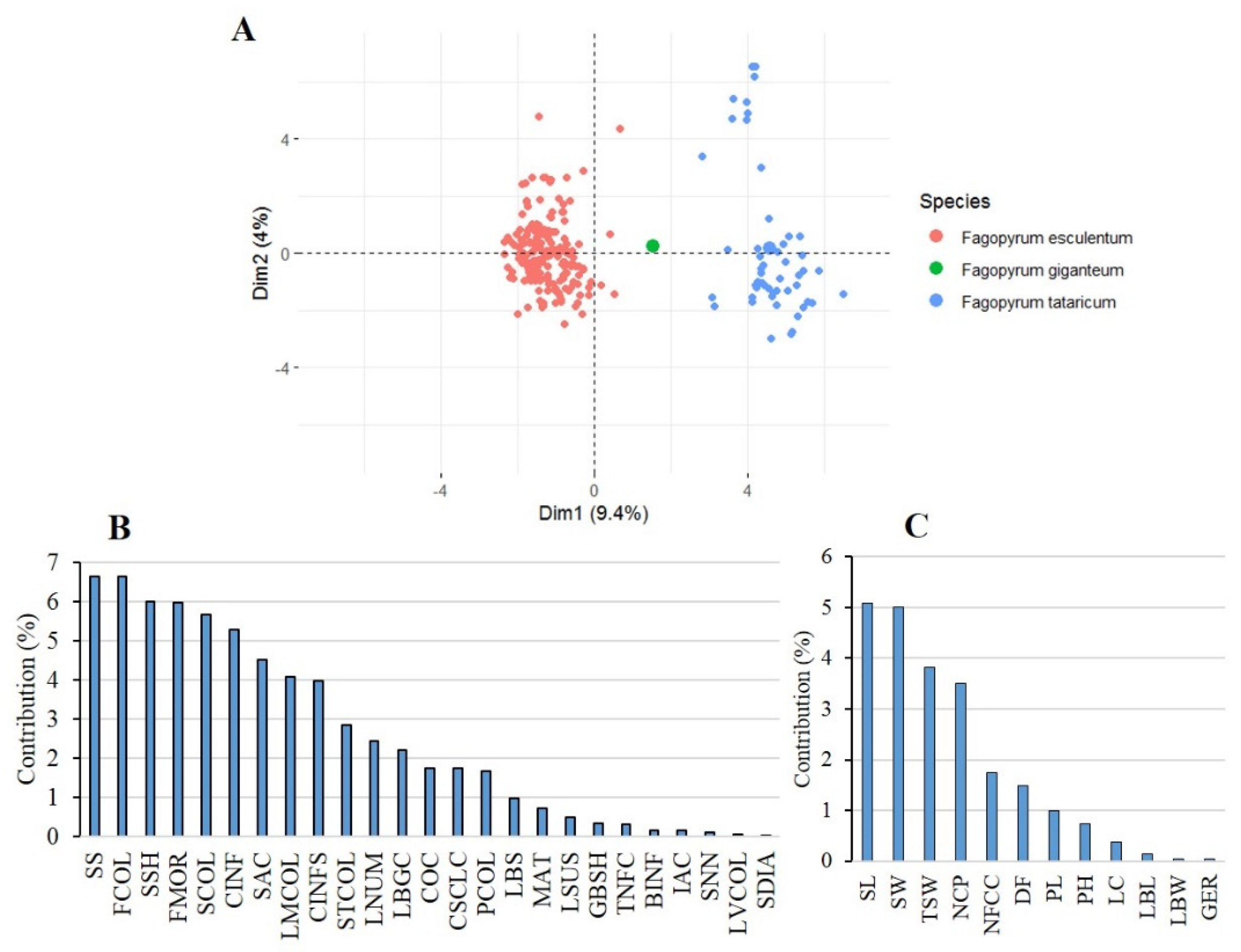
2.1. Patterns of Agro-Morphological Variation and Correlation between Traits
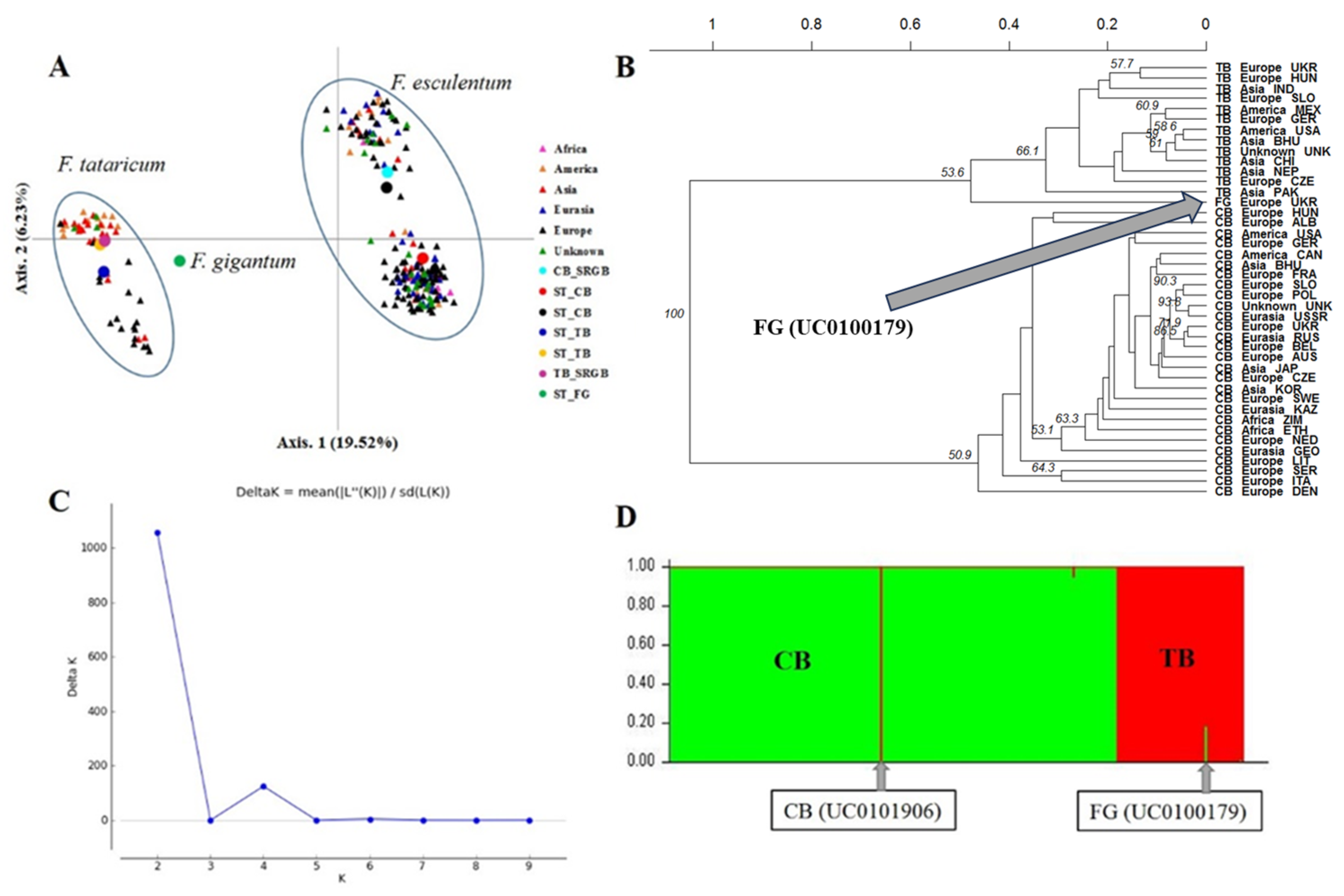
2.2. Patterns of Agro-Morphological Differentiation among and within Species
2.3. Microsatellite Polymorphism and Species-Level Diversity
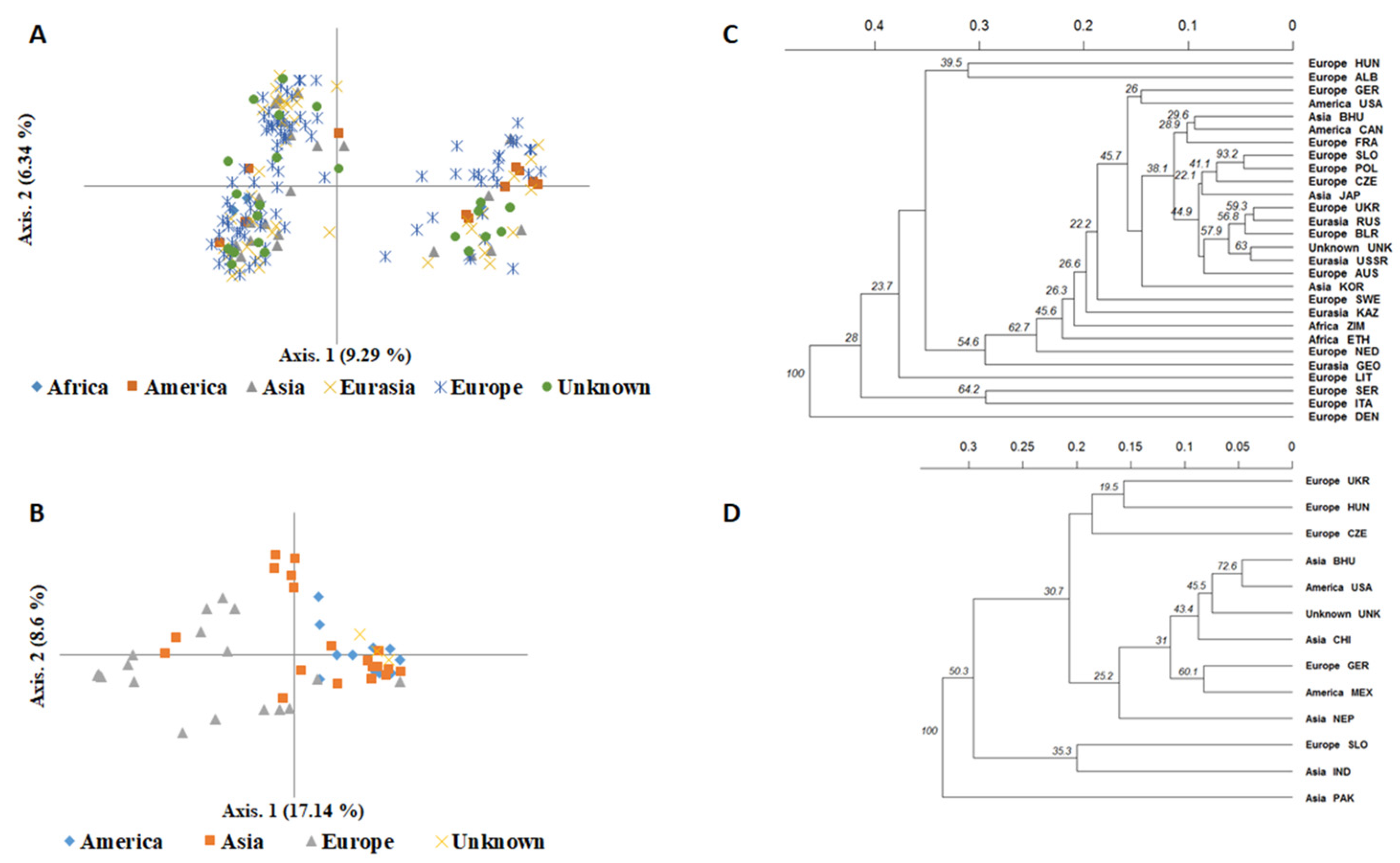
2.4. Genetic Differentiation and Population Structure between and within Species
2.5. Linkage Disequilibrium and Marker-Trait Association Analysis
2.6. Selection of Representative Core Set Collections
3. Discussion
4. Materials and Methods
4.1. Plant Material and Phenotyping Trial
4.2. DNA Extraction and Molecular Analysis
4.3. Data Analysis
4.3.1. Agro-Morphological Diversity
4.3.2. SSR Data Analysis
4.3.3. Linkage Disequilibrium Evaluation and Marker-Traits Associations (MTAs) Analysis
4.3.4. Implementation of the Core Collections
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, K.; He, M.; Fan, Y.; Zhao, H.; Gao, B.; Yang, K.; Li, F.; Tang, Y.; Gao, Q.; Lin, T. Resequencing of global Tartary buckwheat accessions reveals multiple domestication events and key loci associated with agronomic traits. Genome Biol. 2021, 22, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; He, Y.; Zhang, K.; Li, S.; Chen, Y.; He, M.; He, F.; Gao, B.; Yang, D.; Fan, Y. Rewiring of the seed metabolome during Tartary buckwheat domestication. Plant Biotechnol. J. 2023, 21, 150–164. [Google Scholar] [CrossRef]
- Chapman, M.A.; He, Y.; Zhou, M. Beyond a reference genome: Pangenomes and population genomics of underutilized and orphan crops for future food and nutrition security. New Phytol. 2022, 234, 1583–1597. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xu, J.G. Determining the geographical origin of common buckwheat from China by multivariate analysis based on mineral elements, amino acids and vitamins. Sci. Rep. 2017, 7, 9696. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cai, C.; Yao, Y.; Xu, B. Alteration of phenolic profiles and antioxidant capacities of common buckwheat and tartary buckwheat produced in China upon thermal processing. J. Sci. Food Agric. 2019, 99, 5565–5576. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Ma, D.; Li, W.; Xing, L.; Zhang, H.; Wang, Y.; Du, C.; Li, X.; Jia, Z.; Li, X.; et al. High-quality Fagopyrum esculentum genome provides insights into the flavonoid accumulation among different tissues and self-incompatibility. J. Integr. Plant Biol. 2023, 65, 1423–1441. [Google Scholar] [CrossRef]
- Yilmaz, H.Ö.; Ayhan, N.Y.; Meriç, Ç.S. Buckwheat: A useful food and its effects on human health. Curr. Nutr. Food Sci. 2020, 16, 29–34. [Google Scholar] [CrossRef]
- Ruan, J.; Zhou, Y.; Yan, J.; Zhou, M.; Woo, S.-H.; Weng, W.; Cheng, J.; Zhang, K. Tartary buckwheat: An under-utilized edible and medicinal herb for food and nutritional security. Food Rev. Int. 2022, 38, 440–454. [Google Scholar] [CrossRef]
- Zhou, J.; He, W.; Wang, J.; Liao, X.; Xiang, K.; Ma, M.; Liu, Z.; Li, Y.; Tembrock, L.R.; Wu, Z.; et al. The pan-plastome of tartary buckwheat (Fagopyrum tataricum): Key insights into genetic diversity and the history of lineage divergence. BMC Plant Biol. 2023, 23, 212. [Google Scholar] [CrossRef]
- Luthar, Z.; Golob, A.; Germ, M.; Vombergar, B.; Kreft, I. Tartary buckwheat in human nutrition. Plants 2021, 10, 700. [Google Scholar] [CrossRef]
- Zhu, F. Buckwheat starch: Structures, properties, and applications. Trends Food Sci. Technol. 2016, 49, 121–135. [Google Scholar] [CrossRef]
- Singh, M.; Malhotra, N.; Sharma, K. Buckwheat (Fagopyrum sp.) genetic resources: What can they contribute towards nutritional security of changing world? Genet. Resour. Crop Evol. 2020, 67, 1639–1658. [Google Scholar] [CrossRef]
- Joshi, D.C.; Chaudhari, G.V.; Sood, S.; Kant, L.; Pattanayak, A.; Zhang, K.; Fan, Y.; Janovská, D.; Meglič, V.; Zhou, M. Revisiting the versatile buckwheat: Reinvigorating genetic gains through integrated breeding and genomics approach. Planta 2019, 250, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.P.; Rahman, H.; Thushar, S.; Singh, R.K. Healthy and resilient cereals and pseudo-cereals for marginal agriculture: Molecular advances for improving nutrient bioavailability. Front. Genet. 2020, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Baniya, B.K. Present status of buckwheat genetic resources in Nepal. Curr. Adv. Buckwheat Res. 1995, 47–53. Available online: https://citeseerx.ist.psu.edu/document?repid=rep1&type=pdf&doi=448ab40498a265467404074df030f000bac30911 (accessed on 17 March 2023).
- Choi, Y.-M.; Yoon, H.; Lee, S.; Yoon Hyun, D.; Lee, M.C.; Oh, S.; Rauf, M. Characterization of agro-morphological traits of Tartary buckwheat germplasm under spring cultivation and analysis of health-related primary bioactive components in seeds by HPLC method. J. Plant Biol. 2021, 64, 87–98. [Google Scholar] [CrossRef]
- Joshi, B.K.; Okuno, K.; Ohsawa, R.; Hayashi, H.; Otobe, C.; Kawase, M. Characterization and evaluation of Nepalese tartary buckwheat accessions simultaneously in augmented design. Fagopyrum 2011, 28, 23–41. [Google Scholar]
- Rauf, M.; Yoon, H.; Lee, S.; Hyun, D.Y.; Lee, M.C.; Oh, S.; Choi, Y.M. Evaluation of Fagopyrum esculentum Moench germplasm based on agro-morphological traits and the rutin and quercetin content of seeds under spring cultivation. Genet. Resour. Crop Evol. 2020, 67, 1385–1403. [Google Scholar] [CrossRef]
- Kump, B.; Javornik, B. Genetic diversity and relationships among cultivated and wild accessions of tartary buckwheat (Fagopyrum tataricum Gaertn.) as revealed by RAPD markers. Genet. Resour. Crop Evol. 2002, 49, 565–572. [Google Scholar] [CrossRef]
- Kump, B.; Javornik, B. Evaluation of genetic variability among common buckwheat (Fagopyrum esculentum Moench) populations by RAPD markers. Plant Sci. 1996, 114, 149–158. [Google Scholar] [CrossRef]
- Hou, Y.; Zhang, Z.; Wu, B.; Li, Y. Genetic diversity in tartary buckwheat revealed by AFLP analysis. Sci. Agric. Sin. 2009, 42, 4166–4174. [Google Scholar]
- Kishore, G.; Pandey, A.; Dobhal, R.; Gupta, S. Population genetic study of Fagopyrum tataricum from Western Himalaya using ISSR markers. Biochem. Genet. 2013, 51, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Bashir, E.; Mahajan, R.; Mir, R.A.; Dar, W.A.; Zargar, S.M. Unravelling the genetic variability and population structure of buckwheat (Fagopyrum spp.): A collection of north western Himalayas. Nucleus 2021, 64, 93–101. [Google Scholar] [CrossRef]
- Kishore, G.; Gupta, S.; Pandey, A. Assessment of population genetic diversity of Fagopyrum tataricum using SSR molecular marker. Biochem. Syst. Ecol. 2012, 43, 32–41. [Google Scholar] [CrossRef]
- Nazir, M.; Mahajan, R.; Hashim, M.J.; Iqbal, J.; Alyemeni, M.N.; Ganai, B.A.; Zargar, S.M. Deciphering allelic variability and population structure in buckwheat: An analogy between the efficiency of ISSR and SSR markers. Saudi J. Biol. Sci. 2021, 28, 6050–6056. [Google Scholar] [CrossRef]
- Sharma, D.; Nanjundan, J.; Singh, L.; Parmar, N.; Singh, K.H.; Verma, K.S.; Thakur, A.K. Genetic diversity and population structure analysis in Indian mustard germplasm using phenotypic traits and SSR markers. Plant Mol. Biol. Rep. 2022, 40, 579–594. [Google Scholar] [CrossRef]
- Hou, S.; Ren, X.; Yang, Y.; Wang, D.; Du, W.; Wang, X.; Li, H.; Han, Y.; Liu, L.; Sun, Z. Genome-wide development of polymorphic microsatellite markers and association analysis of major agronomic traits in core germplasm resources of tartary buckwheat. Front. Plant Sci. 2022, 13, 357. [Google Scholar] [CrossRef]
- Mascher, M.; Schreiber, M.; Scholz, U.; Graner, A.; Reif, J.C.; Stein, N. Genebank genomics bridges the gap between the conservation of crop diversity and plant breeding. Nat. Genet. 2019, 51, 1076–1081. [Google Scholar] [CrossRef]
- Song, Y.; Cheng, Z.; Dong, Y.; Liu, D.; Bai, K.; Jarvis, D.; Feng, J.; Long, C. Diversity of Tartary buckwheat (Fagopyrum tataricum) landraces from Liangshan, Southwest China: Evidence from morphology and SSR markers. Agronomy 2022, 12, 1022. [Google Scholar] [CrossRef]
- Bhat, S.; Nazir, M.; Zargar, S.A.; Naik, S.; Dar, W.A.; Bhat, B.A.; Mahajan, R.; Ganai, B.A.; Sofi, P.A.; Zargar, S.M. In-depth morphological assessment revealed significant genetic variability in common buckwheat (Fagopyrum esculentum) and tartary buckwheat (Fagopyrum tataricum) germplasm. Plant Genet. Res. 2023, 20, 417–424. [Google Scholar] [CrossRef]
- Misra, A.K.; Roy, S.; Singh, S.K.; Rathi, R.S.; Harish, G.D. Morphological diversity of buckwheat (Fagopyrum spp.) landraces from Northeast India. Indian J. Plant Genet. Res. 2019, 32, 11–17. [Google Scholar] [CrossRef]
- Naik, S.; Mahajan, R.; Sofi, P.A.; Abidi, I.; Ali, G.; Nehvi, F.A.; Khan, I.; Bhat, S.A.; Bhat, M.A.; Bhat, B.A. Characterisation of buckwheat (Fagopyrum spp.) diversity of the northwestern Himalayas. Crop Pasture Sci. 2022. [Google Scholar] [CrossRef]
- Joshi, B.K.; Okuno, K.; Ohsawa, R.; Bimb, H.P. Principal component and cluster analyses of Nepalese Tartary buckwheat diversity. Fagopyrum 2010, 27, 55–66. [Google Scholar]
- Park, J.E.; Kang, Y.; Han, G.D.; Yildiz, M.; Kim, S.H.; Kim, C.; Chung, Y.S. Diversity study of common buckwheat germplasm in the Republic of Korea using GBS. Plant Biotechnol. Rep. 2022, 16, 799–803. [Google Scholar] [CrossRef]
- Song, J.Y.; Lee, G.A.; Yoon, M.S.; Ma, K.H.; Choi, Y.M.; Lee, J.R.; Jung, Y.J.; Park, H.J.; Kim, C.K.; Lee, M.C. Analysis of Genetic Diversity and Population Structure of Buckwheat (Fagopyrum esculentum Moench.) Landraces of Korea Using SSR Markers. In Buckwheat Germplasm in the World; Zhou, M., Kreft, I., Suvorova, G., Tang, Y., Woo, S.H., Eds.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherland, 2018; pp. 315–331. [Google Scholar]
- Janovská, D.; Jágr, M.; Svoboda, P.; Dvořáček, V.; Meglič, V.; Hlásná Čepková, P. Breeding buckwheat for nutritional quality in the Czech Republic. Plants 2021, 10, 1262. [Google Scholar] [CrossRef]
- Gupta, N.; Sharma, S.K.; Rana, J.C.; Chauhan, R.S. AFLP fingerprinting of tartary buckwheat accessions (Fagopyrum tataricum) displaying rutin content variation. Fitoterapia 2012, 83, 1131–1137. [Google Scholar] [CrossRef]
- Hou, S.; Sun, Z.; Linghu, B.; Xu, D.; Wu, B.; Zhang, B.; Wang, X.; Han, Y.; Zhang, L.; Qiao, Z. Genetic diversity of buckwheat cultivars (Fagopyrum tartaricum Gaertn.) assessed with SSR markers developed from genome survey sequences. Plant Mol. Biol. Rep. 2016, 34, 233–241. [Google Scholar] [CrossRef]
- Nay, M.M.; Byrne, S.L.; Pérez, E.A.; Walter, A.; Studer, B. Genetic characterization of buckwheat accessions through genome-wide allele-frequency fingerprints. Folia Biol. Geol. 2020, 61, 17–23. [Google Scholar] [CrossRef]
- Joshi, B.K. Correlation, regression and path coefficient analyses for some yield components in common and Tartary buckwheat in Nepal. Fagopyrum 2005, 22, 77–82. [Google Scholar]
- Dar, F.A.; Tahir, I.; Rehman, R.U. Morphological characterization reveals high intraspecies diversity in Fagopyrum esculentum Moench and Fagopyrum sagittatum Gilib from north-western Himalayan regions. Agric. Res. 2022, 11, 398–409. [Google Scholar] [CrossRef]
- Kapoor, C.; Avasthe, R.K.; Chettri, P.K.; Gopi, R.; Kalita, H.; Rana, J.C. Multivariate analysis to evaluate common and tartary buckwheat germplasm in Sikkim. Indian J. Plant Genet. Res. 2018, 31, 134–141. [Google Scholar] [CrossRef]
- Tetsuka, T.; Uchino, A. Variation in seed shape and husk color in Japanese native cultivars of common buckwheat (Fagopyrum esculentum Moench). Plant Prod. Sci. 2005, 8, 60–64. [Google Scholar] [CrossRef][Green Version]
- Janovska, D.; Stehno, Z.; Cepkova, P. Evaluation of Common Buckwheat Genetic Resources in Czech Gene Bank. In Advances in Buckwheat Research, Proceedings of the 10th International Symposium on Buckwheat, Yangling, China, 14–18 August 2007; Northwest A&F University Press: Yangling, China, 2007; pp. 31–40. [Google Scholar]
- Stehno, Z.; Janovska, D.; Wang, Z. Comparison of Selected Traits of Common and Tartary Buckwheat Originated from China and Czech Republic under Conditions of the Czech Republic. In Advances in Buckwheat Research, Proceedings of the 10th International Symposium on Buckwheat, Yangling, China, 14–18 August 2007; Northwest A&F University Press: Yangling, China, 2007; pp. 252–256. [Google Scholar]
- Chrungoo, N.K.; Dohtdong, L.; Chettry, U. Genome Plasticity in Buckwheat. In Gene Pool Diversity and Crop Improvement Sustainable Development and Biodiversity; Rajpal, V.R., Rao, S.R., Raina, S.N., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 227–239. [Google Scholar] [CrossRef]
- Facho, Z.H.; Farhatullah, I.H.K.; Khan, N.U.; Ali, S. Morphological characterization and estimation of genotype × environment interaction of indigenous buckwheat germplasm collected from Gilgit Baltistan Pakistan. Pak. J. Bot. 2016, 48, 2391–2398. Available online: https://www.researchgate.net/publication/315708780_Morphological_characterization_and_estimation_of_genotype_environment_interaction_of_indigenous_buckwheat_germplasm_collected_from_Gilgit_Baltistan_Pakistan (accessed on 26 May 2023).
- Hunt, H.V.; Shang, X.; Jones, M.K. Buckwheat: A crop from outside the major Chinese domestication centres? A review of the archaeobotanical, palynological and genetic evidence. Veg. Hist. Archaeobot. 2018, 27, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.K.; Baniya, B.K. A diversity in qualitative traits of Nepalese cultivated buckwheat species. Fagopyrum 2006, 23, 23–27. [Google Scholar]
- Oomah, B.D.; Campbell, C.G.; Mazza, G. Effects of cultivar and environment on phenolic acids in buckwheat. Euphytica 1996, 90, 73–77. [Google Scholar] [CrossRef]
- Saunders Bulan, M.; Wu, J.; Emshwiller, E.; Berres, M.E.; Posner, J.L.; Peng, D.; Wang, X.; Li, J.; Stoltenberg, D.E.; Zhang, Y. Social and environmental influences on tartary buckwheat (Fagopyrum tataricum Gaertn.) varietal diversity in Yunnan, China. Genet. Resour. Crop Evol. 2017, 64, 113–125. [Google Scholar] [CrossRef]
- Cobos, M.J.; Rubio, J.; Fernández-Romero, M.D.; Garza, R.; Moreno, M.T.; Millán, T.; Gil, J. Genetic analysis of seed size, yield and days to flowering in a chickpea recombinant inbred line population derived from a Kabuli × Desi cross. Ann. Appl. Biol. 2007, 151, 33–42. [Google Scholar] [CrossRef]
- Matsui, K.; Yasui, Y. Genetic and genomic research for the development of an efficient breeding system in heterostylous self-incompatible common buckwheat (Fagopyrum esculentum). Theor. Appl. Genet. 2020, 133, 1641–1653. [Google Scholar] [CrossRef]
- Senthilkumaran, R.; Bisht, I.S.; Bhat, K.V.; Rana, J.C. Diversity in buckwheat (Fagopyrum spp.) landrace populations from north-western Indian Himalayas. Genet. Resour. Crop Evol. 2008, 55, 287–302. [Google Scholar] [CrossRef]
- Shukla, A.; Srivastava, N.; Suneja, P.; Yadav, S.K.; Hussain, Z.; Rana, J.C.; Yadav, S. Genetic diversity analysis in Buckwheat germplasm for nutritional traits. Indian J. Exp. Biol. 2018, 56, 827–837. [Google Scholar]
- Guijarro-Real, C.; Prohens, J.; Rodríguez-Burruezo, A.; Fita, A. Potential of wall rocket (Diplotaxis erucoides) as a new crop: Influence of the growing conditions on the visual quality of the final product. Sci. Hort. 2019, 258, 108778. [Google Scholar] [CrossRef]
- Jeon, J.; Kim, J.K.; Wu, Q.; Park, S.U. Effects of cold stress on transcripts and metabolites in tartary buckwheat (Fagopyrum tataricum). Environ. Exp. Bot. 2018, 155, 488–496. [Google Scholar] [CrossRef]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef]
- Debnath, N.R.; Rasul, M.G.; Sarker, M.M.H.; Rahman, M.H.; Paul, A.K. Genetic divergence in buckwheat (Fagopyrum esculentum Moench.). Int. J. Sustain. Crop Prod. 2008, 3, 60–68. [Google Scholar]
- Li, J.; Feng, S.; Qu, Y.; Gong, X.; Luo, Y.; Yang, Q.; Zhang, Y.; Dang, K.; Gao, X.; Feng, B. Identifying the primary meteorological factors affecting the growth and development of Tartary buckwheat and a comprehensive landrace evaluation using a multi-environment phenotypic investigation. J. Sci. Food Agric. 2021, 101, 6104–6116. [Google Scholar] [CrossRef]
- Ambreen, H.; Kumar, S.; Kumar, A.; Agarwal, M.; Jagannath, A.; Goel, S. Association mapping for important agronomic traits in safflower (Carthamus tinctorius L.) core collection using microsatellite markers. Front. Plant Sci. 2018, 9, 402. [Google Scholar] [CrossRef]
- Soren, K.R.; Konda, A.K.; Gangwar, P.; Tiwari, V.A.; Shanmugavadivel, P.S.; Parihar, A.K.; Dixit, G.P.; Singh, N.P. Development of SSR markers and association studies of markers with phenology and yield-related traits in grass pea (Lathyrus sativus). Crop Pasture Sci. 2020, 71, 768–775. [Google Scholar] [CrossRef]
- Iwata, H.; Imon, K.; Tsumura, Y.; Ohsawa, R. Genetic diversity among Japanese indigenous common buckwheat (Fagopyrum esculentum) cultivars as determined from amplified fragment length polymorphism and simple sequence repeat markers and quantitative agronomic traits. Genome 2005, 48, 367–377. [Google Scholar] [CrossRef]
- Konishi, T.; Iwata, H.; Yashiro, K.; Tsumura, Y.; Ohsawa, R.; Yasui, Y.; Ohnishi, O. Development and characterization of microsatellite markers for common buckwheat. Breed. Sci. 2006, 56, 277–285. [Google Scholar] [CrossRef]
- Ma, K.H.; Kim, N.S.; Lee, G.A.; Lee, S.Y.; Lee, J.K.; Yi, J.Y.; Park, Y.J.; Kim, T.S.; Gwag, J.G.; Kwon, S.J. Development of SSR markers for studies of diversity in the genus Fagopyrum. Theor. Appl. Genet. 2009, 119, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Li, R.; Chen, Q.; Li, Y.; Pan, F.; Chen, Q. De novo sequencing of seed transcriptome and development of genic-SSR markers in common buckwheat (Fagopyrum esculentum). Mol. Breed. 2017, 37, 147. [Google Scholar] [CrossRef]
- Fang, X.; Huang, K.; Nie, J.; Zhang Yuanli Zhang Yuke Li, Y.; Wang, W.; Xu, X.; Ruan, R.; Yuan, X. Genome-wide mining, characterization, and development of microsatellite markers in Tartary buckwheat (Fagopyrum tataricum Garetn.). Euphytica 2019, 215, 183. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, Z.; Wu, B. Construction and application of SSR molecular markers system for genetic diversity analysis of Chinese tartary buckwheat germplasm resources. Sci. Agric. Sin. 2012, 45, 1042–1053. [Google Scholar]
- Grahić, J.; Okić, A.; Šimon, S.; Djikić, M.; Gadžo, D.; Pejić, I.; Gaši, F. Genetic relationships and diversity of common buckwheat accessions in Bosnia and Herzegovina. Agronomy 2022, 12, 2676. [Google Scholar] [CrossRef]
- Rout, A.; Chrungoo, N.K. Genetic variation and species relationships in Himalayan buckwheats as revealed by SDS PAGE of endosperm proteins extracted from single seeds and RAPD based DNA fingerprints. Genet. Resour. Crop Evol. 2007, 54, 767–777. [Google Scholar] [CrossRef]
- Facho, Z.H.; Farhatullah, A.; Ali, S. Species divergence and diversity in buckwheat landraces collected from the western Himalayan region of Pakistan. Pak. J. Bot. 2019, 51, 2215–2224. [Google Scholar] [CrossRef]
- Barcaccia, G.; Volpato, M.; Gentili, R.; Abeli, T.; Galla, G.; Orsenigo, S.; Citterio, S.; Sgorbati, S.; Rossi, G. Genetic identity of common buckwheat (Fagopyrum esculentum Moench) landraces locally cultivated in the Alps. Genet. Resour. Crop Evol. 2016, 63, 639–651. [Google Scholar] [CrossRef]
- Khazaei, H.; Fedoruk, M.; Caron, C.T.; Vandenberg, A.; Bett, K.E. Single nucleotide polymorphism markers associated with seed quality characteristics of cultivated lentil. J. Plant Genome 2018, 11, 170051. [Google Scholar] [CrossRef]
- Shi, T.X.; Li, R.Y.; Zheng, R.; Chen, Q.F.; Li, H.Y.; Huang, J.; Zhu, L.W.; Liang, C.G. Mapping QTLs for 1000-grain weight and genes controlling hull type using SNP marker in Tartary buckwheat (Fagopyrum tataricum). BMC Genom. 2021, 22, 142. [Google Scholar] [CrossRef]
- IPGRI Descriptors for Buckwheat (Fagopyrum spp.). 1994, pp. 1–48. Available online: https://cgspace.cgiar.org/handle/10568/72849 (accessed on 16 May 2021).
- UPOV Buckwheat Fagopyrum Esculentum Moench. Guidelines for the Conduct of Tests for Distinctness, Uniformity and Stability. 2012, pp. 1–27. Available online: https://www.upov.int/edocs/tgdocs/en/tg278.pdf (accessed on 16 May 2021).
- Pipan, B.; Zupančič, M.; Blatnik, E.; Dolničar, P.; Meglič, V. Comparison of six genomic DNA extraction methods for molecular downstream applications of apple tree (Malus × domestica). Cogent Food Agric. 2018, 4, 1540094. [Google Scholar] [CrossRef]
- Pipan, B.; Meglič, V. Diversification and genetic structure of the western-to-eastern progression of European Phaseolus vulgaris L. germplasm. BMC Plant Biol. 2019, 19, 442. [Google Scholar] [CrossRef] [PubMed]
- Grosjean, P.; Ibanez, F.; Etienne, M.; Grosjean, M.P. Package ‘Pastecs’. 2018. Available online: http://masterdistfiles.gentoo.org/pub/cran/web/packages/pastecs/pastecs.pdf (accessed on 17 March 2023).
- Guevara, M.R.; Hartmann, D.; Mendoza, M. Diverse: An R package to analyze diversity in complex systems. R J. 2016, 8, 60. Available online: https://svn.r-project.org/Rjournal/html/archive/2016-2/guevara-hartmann-mendoza.pdf (accessed on 17 March 2023). [CrossRef]
- Shannon, C.E.; Weaver, W. A Mathematical Model of Communication; University of Illinois Press: Urbana, IL, USA, 1949; pp. 11–20. [Google Scholar]
- Taiyun, W.; Viliam, S. R package “corrplot”: Visualization of a Correlation Matrix (Version 0.84). Statistician 2017, 56, e24. [Google Scholar]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses; R Package Version 1.0.3. 2020. Available online: https://CRAN.R-project.org/package=factoextra (accessed on 17 March 2023).
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef]
- Goudet, J. HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef]
- Dray, S.; Siberchicot, M.A. Package ‘ade4’; Université de Lyon: Lyon, France, 2017; Available online: http://cran.nexr.com/web/packages/ade4/ade4.pdf (accessed on 16 March 2023).
- Peakall, R.O.D.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A.; Von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Chung, H.K.; Cho, G.T.; Ma, K.H.; Chandrabalan, D.; Gwag, J.G.; Kim, T.S.; Cho, E.G.; Park, Y.J. PowerCore: A program applying the advanced M strategy with a heuristic search for establishing core sets. Bioinformatics 2007, 23, 2155–2162. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | F. esculentum | F. tataricum | Differences | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Min | Max | Mean ± SE | CV (%) | Min | Max | Mean ± SE | CV (%) | ||
| Germination period (days) | 5.00 | 11.00 | 7.40 ± 0.09 | 16.88 | 7.00 | 11.00 | 7.54 ± 0.15 | 14.01 | NS |
| Days to flowering | 45.00 | 66.00 | 54.07 ± 0.28 | 7.26 | 51.00 | 66.00 | 58.38 ± 0.5 | 6.26 | *** |
| Plant height (cm) | 57.60 | 143.00 | 105.18 ± 1.12 | 14.70 | 64.00 | 123.00 | 92.98 ± 2.16 | 16.73 | *** |
| Number of flower clusters per cyme | 1.80 | 17.60 | 10.59 ± 0.2 | 26.07 | 1.60 | 20.80 | 13.98 ± 0.5 | 26.09 | *** |
| Number of cymes per plant | 3.60 | 9.00 | 6.07 ± 0.07 | 16.89 | 5.80 | 12.80 | 8.56 ± 0.19 | 15.71 | *** |
| Length of cyme (cm) | 18.80 | 92.00 | 59.5 ± 0.83 | 19.40 | 17.00 | 93.00 | 64.08 ± 1.79 | 20.25 | ** |
| Petiole length (mm) | 4.33 | 11.57 | 7.05 ± 0.09 | 17.90 | 5.45 | 11.25 | 8.11 ± 0.16 | 14.39 | *** |
| Leaf blade length (cm) | 5.22 | 11.20 | 7.97 ± 0.08 | 13.92 | 5.58 | 9.86 | 7.46 ± 0.14 | 13.58 | *** |
| Leaf blade width (cm) | 5.52 | 12.64 | 8.39 ± 0.08 | 13.84 | 5.22 | 10.64 | 8.50 ± 0.20 | 17.23 | NS |
| Seed length (mm) | 4.43 | 7.23 | 6.11 ± 0.03 | 6.63 | 3.20 | 6.16 | 4.68 ± 0.07 | 11.59 | *** |
| Seed width (mm) | 2.85 | 5.44 | 4.18 ± 0.03 | 10.45 | 1.98 | 4.55 | 2.99 ± 0.05 | 11.07 | *** |
| 1000-seed weight (g) | 13.56 | 38.70 | 25.42 ± 0.33 | 18.15 | 9.00 | 26.27 | 16.2 ± 0.45 | 20.02 | *** |
| Marker | F. esculentum | F. tataricum | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MAF | Ar | Na | Ho | He | Fis | PIC | MAF | Ar | Na | Ho | He | PIC | Fis | |
| Fem1303 | 0.65 | 8.21 | 12.00 | 0.55 | 0.54 | −0.03 | 0.50 | 1.00 | 1.00 | 1.00 | 0.00 | 0.00 | 0.00 | NA |
| Fem1322 | 0.18 | 10.67 | 16.00 | 0.98 | 0.89 | −0.11 | 0.88 | 0.61 | 3.00 | 3.00 | 0.59 | 0.53 | 0.46 | −0.10 |
| Fem1407 | 0.37 | 11.60 | 20.00 | 0.91 | 0.80 | −0.13 | 0.78 | 0.43 | 6.84 | 7.00 | 0.71 | 0.70 | 0.65 | −0.01 |
| Fem1528 | 0.19 | 13.93 | 18.00 | 0.92 | 0.88 | −0.05 | 0.87 | 0.31 | 6.99 | 7.00 | 0.98 | 0.79 | 0.76 | −0.24 |
| Fem1840 | 0.29 | 11.45 | 15.00 | 0.80 | 0.84 | 0.05 | 0.82 | 0.24 | 9.00 | 9.00 | 0.60 | 0.83 | 0.81 | 0.29 |
| SXAU048 | 0.21 | 15.47 | 25.00 | 0.83 | 0.88 | 0.05 | 0.87 | 0.41 | 5.00 | 5.00 | 0.84 | 0.73 | 0.70 | −0.15 |
| SXAU060 | 0.51 | 7.34 | 14.00 | 0.63 | 0.69 | 0.09 | 0.66 | 0.63 | 2.00 | 2.00 | 0.00 | 0.47 | 0.36 | 1.00 |
| SXAU089 | 0.21 | 12.00 | 17.00 | 0.53 | 0.90 | 0.27 | 0.89 | 0.34 | 5.00 | 5.00 | 1.00 | 0.74 | 0.69 | −0.36 |
| SXAU129 | 0.23 | 10.50 | 17.00 | 0.94 | 0.89 | −0.06 | 0.88 | 0.58 | 4.92 | 5.00 | 0.75 | 0.57 | 0.50 | −0.31 |
| SXAU138 | 0.26 | 7.48 | 14.00 | 0.92 | 0.84 | −0.10 | 0.82 | 0.54 | 4.88 | 5.00 | 0.78 | 0.53 | 0.43 | −0.47 |
| Ft1_114 | 0.26 | 7.52 | 11.00 | 0.90 | 0.83 | −0.09 | 0.81 | 0.44 | 8.76 | 9.00 | 0.96 | 0.70 | 0.65 | −0.38 |
| Ft2_1743 | 0.33 | 6.91 | 9.00 | 0.96 | 0.78 | −0.23 | 0.75 | 0.50 | 2.99 | 3.00 | 0.96 | 0.52 | 0.40 | −0.85 |
| Ft3_572 | 0.24 | 9.72 | 13.00 | 0.98 | 0.87 | −0.12 | 0.86 | 0.22 | 13.92 | 14.00 | 0.90 | 0.89 | 0.88 | −0.02 |
| Ft4_2725 | 0.24 | 7.55 | 13.00 | 0.86 | 0.81 | −0.07 | 0.78 | 0.25 | 10.00 | 10.00 | 1.00 | 0.86 | 0.85 | −0.16 |
| Ft5_2899 | 0.32 | 6.77 | 13.00 | 0.95 | 0.81 | −0.18 | 0.79 | 0.20 | 13.99 | 14.00 | 0.96 | 0.90 | 0.89 | −0.07 |
| Ft6_2849 | 0.42 | 5.64 | 13.00 | 0.82 | 0.73 | −0.14 | 0.69 | 0.33 | 7.99 | 8.00 | 0.96 | 0.75 | 0.71 | −0.29 |
| Ft7_382 | 0.39 | 9.46 | 16.00 | 0.92 | 0.80 | −0.15 | 0.78 | 0.49 | 5.92 | 6.00 | 0.45 | 0.67 | 0.62 | 0.33 |
| Ft8_605 | 0.56 | 10.95 | 16.00 | 0.70 | 0.65 | −0.07 | 0.64 | 0.31 | 7.99 | 8.00 | 1.00 | 0.79 | 0.76 | −0.27 |
| GB_FE_025 | 0.47 | 6.35 | 13.00 | 0.98 | 0.69 | −0.42 | 0.65 | 0.46 | 5.00 | 5.00 | 0.94 | 0.64 | 0.57 | −0.48 |
| GB_FE_035 | 0.48 | 4.52 | 9.00 | 1.00 | 0.57 | −0.75 | 0.48 | 0.44 | 6.99 | 7.00 | 1.00 | 0.69 | 0.64 | −0.45 |
| GB_FE_121 | 0.41 | 7.69 | 12.00 | 0.99 | 0.68 | −0.45 | 0.63 | 0.54 | 3.00 | 3.00 | 0.92 | 0.56 | 0.48 | −0.64 |
| GB_FE_054 | 0.23 | 5.96 | 14.00 | 0.94 | 0.84 | −0.12 | 0.82 | 0.32 | 7.00 | 7.00 | 0.90 | 0.80 | 0.77 | −0.13 |
| TBP_5 | 0.35 | 6.90 | 10.00 | 0.56 | 0.78 | 0.29 | 0.75 | 0.47 | 5.00 | 5.00 | 0.53 | 0.68 | 0.63 | 0.22 |
| TBP_6 | 0.63 | 2.78 | 6.00 | 0.06 | 0.51 | 0.89 | 0.43 | 0.64 | 4.92 | 5.00 | 0.06 | 0.53 | 0.47 | 0.89 |
| Mean | 0.35 | 8.64 | 13,95 | 0.82 | 0.77 | −0.07 | 0.74 | 0,44 | 6.34 | 6.38 | 0.74 | 0.66 | 0.61 | −0.12 |
| N | Ar | Na | Ho | He | Fis | ||
|---|---|---|---|---|---|---|---|
| F. esculentum | Africa | 3 | 3.66 | 3.67 | 0.75 | 0.64 | −0.15 |
| America | 11 | 3.78 | 6.92 | 0.81 | 0.74 | −0.10 | |
| Asia | 22 | 3.73 | 8.75 | 0.80 | 0.74 | −0.07 | |
| Eurasia | 40 | 3.71 | 9.46 | 0.84 | 0.76 | −0.11 | |
| Europe | 92 | 3.80 | 12.71 | 0.82 | 0.77 | −0.07 | |
| Unknown | 22 | 3.81 | 8.75 | 0.83 | 0.76 | −0.09 | |
| Mean | 3.75 | 8.38 | 0.81 | 0.74 | −0.10 | ||
| Core collection | 23 | 8.63 | 9.21 | 0.83 | 0.78 | −0.06 | |
| F. tataricum | Africa | 0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| America | 12 | 2.81 | 4.17 | 0.72 | 0.57 | −0.26 | |
| Asia | 19 | 2.99 | 4.92 | 0.76 | 0.63 | −0.17 | |
| Eurasia | 0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| Europe | 18 | 3.09 | 5.75 | 0.73 | 0.66 | −0.11 | |
| Unknown | 3 | 2.75 | 2.75 | 0.79 | 0.52 | −0.55 | |
| Mean | 1.94 | 3.52 | 0.60 | 0.48 | −0.22 | ||
| Core collection | 13 | 5.53 | 5.63 | 0.73 | 0.67 | −0.09 | |
| Species | F | 3.706 | 9.159 | 28.028 | 3.986 | 8.425 | 0.855 |
| p | 0.112 | 0.029 | 0.003 | 0.102 | 0.034 | 0.398 | |
| Region | F | 1.584 | 1.135 | 3.738 | 1.119 | 1.443 | 0.786 |
| p | 0.313 | 0.446 | 0.087 | 0.453 | 0.349 | 0.601 | |
| DF | Sum of Squares | Mean Square | Variance Components | Total Variance (%) | p | |
|---|---|---|---|---|---|---|
| Buckwheat collection | ||||||
| Variations between species | 1 | 920.35 | 920.35 | 5.36 | 23.83 | 0.001 |
| Between accessions within species | 240 | 2019.98 | 8.42 | 0.00 | 0.00 | 1.000 |
| Within accessions | 242 | 2326.50 | 9.61 | 9.61 | 76.17 | 0.001 |
| F. esculentum | ||||||
| Variations between regions | 5 | 93.58 | 18.71 | 0.00 | 0.00 | 0.930 |
| Variations between countries within the region | 22 | 437.47 | 19.88 | 0.38 | 2.21 | 1 |
| Between accessions within the country | 162 | 2523.03 | 15.57 | 0.00 | 0.00 | 1 |
| Within accessions | 190 | 3631.01 | 19.11 | 19.11 | 97.79 | 0.0001 |
| F. tataricum | ||||||
| Variations between regions | 3 | 99.92 | 33.30 | 0.38 | 2.45 | 0.220 |
| Variations between countries within the region | 9 | 149.16 | 16.57 | 1.01 | 6.53 | 1 |
| Between accessions within the country | 39 | 429.35 | 11.00 | 0.00 | 0.00 | 1 |
| Within accessions | 52 | 895.30 | 17.21 | 17.21 | 91.02 | 0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pipan, B.; Sinkovič, L.; Neji, M.; Janovská, D.; Zhou, M.; Meglič, V. Agro-Morphological and Molecular Characterization Reveal Deep Insights in Promising Genetic Diversity and Marker-Trait Associations in Fagopyrum esculentum and Fagopyrum tataricum. Plants 2023, 12, 3321. https://doi.org/10.3390/plants12183321
Pipan B, Sinkovič L, Neji M, Janovská D, Zhou M, Meglič V. Agro-Morphological and Molecular Characterization Reveal Deep Insights in Promising Genetic Diversity and Marker-Trait Associations in Fagopyrum esculentum and Fagopyrum tataricum. Plants. 2023; 12(18):3321. https://doi.org/10.3390/plants12183321
Chicago/Turabian StylePipan, Barbara, Lovro Sinkovič, Mohamed Neji, Dagmar Janovská, Meiliang Zhou, and Vladimir Meglič. 2023. "Agro-Morphological and Molecular Characterization Reveal Deep Insights in Promising Genetic Diversity and Marker-Trait Associations in Fagopyrum esculentum and Fagopyrum tataricum" Plants 12, no. 18: 3321. https://doi.org/10.3390/plants12183321
APA StylePipan, B., Sinkovič, L., Neji, M., Janovská, D., Zhou, M., & Meglič, V. (2023). Agro-Morphological and Molecular Characterization Reveal Deep Insights in Promising Genetic Diversity and Marker-Trait Associations in Fagopyrum esculentum and Fagopyrum tataricum. Plants, 12(18), 3321. https://doi.org/10.3390/plants12183321