

Bridging the Gap: Combining Genomics and Transcriptomics Approaches to Understand Stylosanthes scabra, an Orphan Legume from the Brazilian Caatinga

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Plant Material and Genomic DNA Extraction
2.2. Sample Sequencing and Genome Size Estimation by Flow Cytometry
2.3. S. scabra De Novo Genome Assembly and BUSCO Analysis
2.4. Gene Prediction, Functional Annotations and General Gene Features
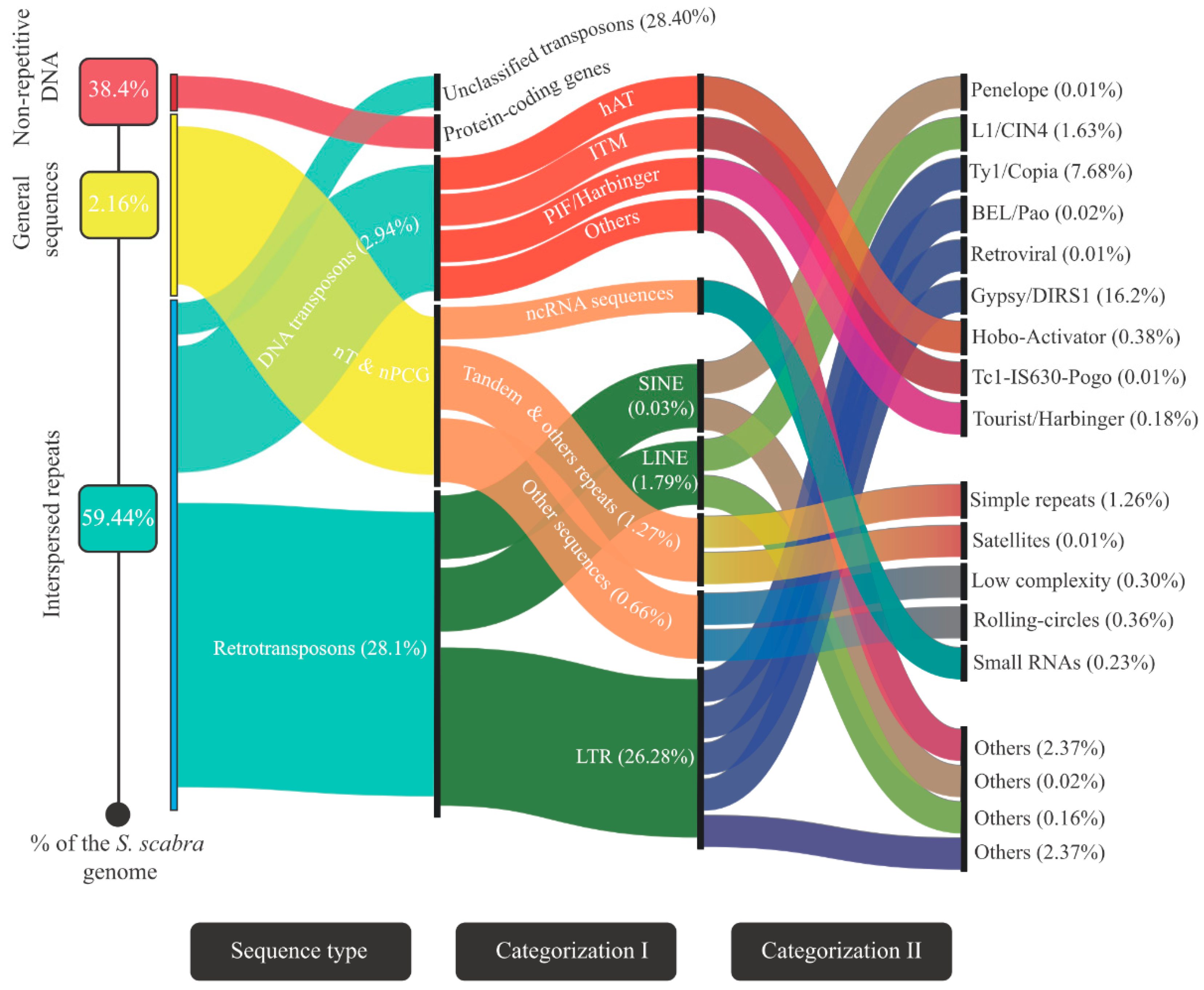
2.5. Transposable Elements and Other Repetitive Sequences’ Mining and Annotation
2.6. Gene Family Identification and Respective Expansion/Contraction Analysis
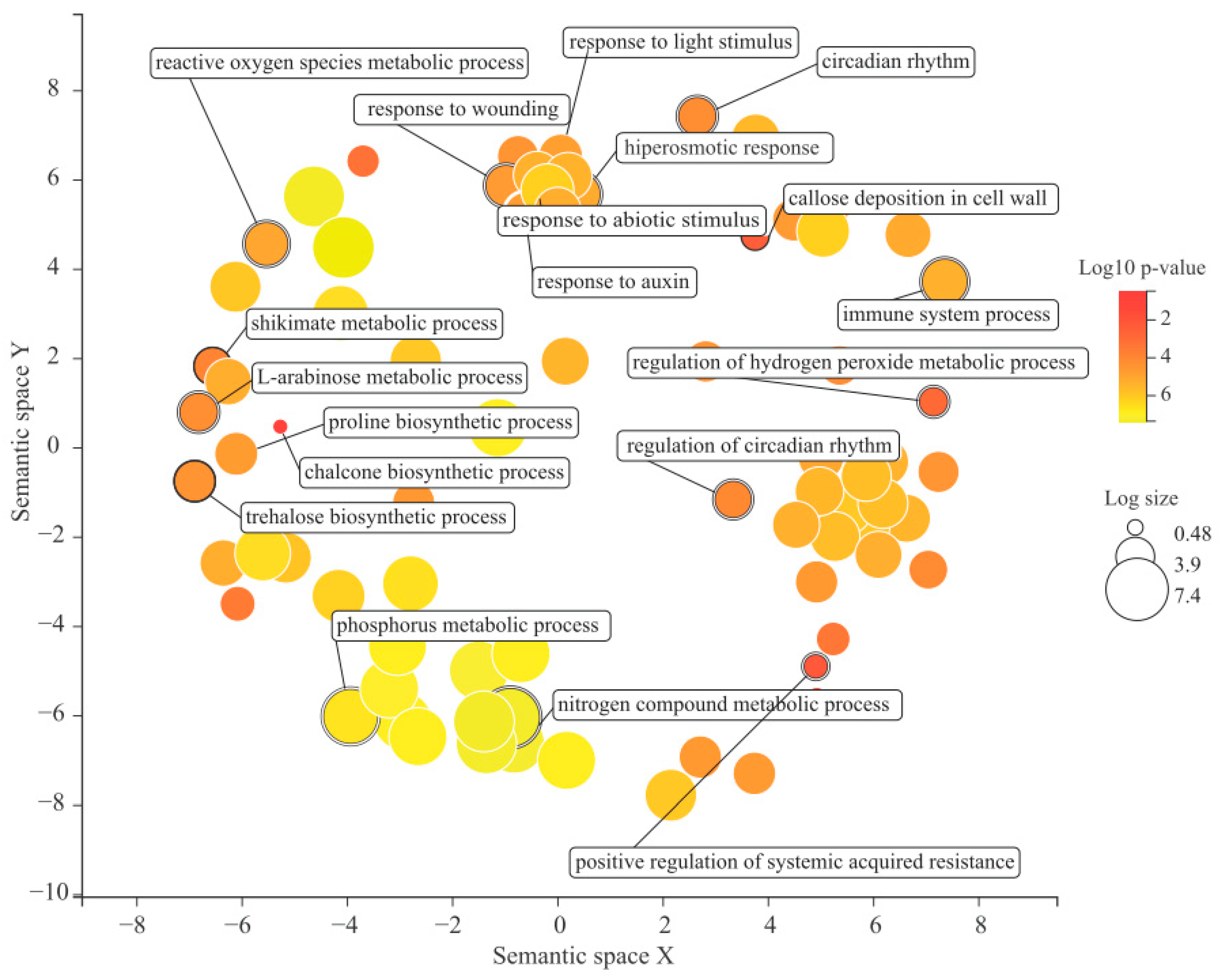
2.7. Gene Ontology Enrichment Analysis
2.8. “R” and “PRR” Gene Mining and Identification
2.9. In Silico Anchoring of S. scabra “R” and “PRR” Genes in Soybean QTL Regions Associated with Resistance to the Phakopsora pachyrhizi
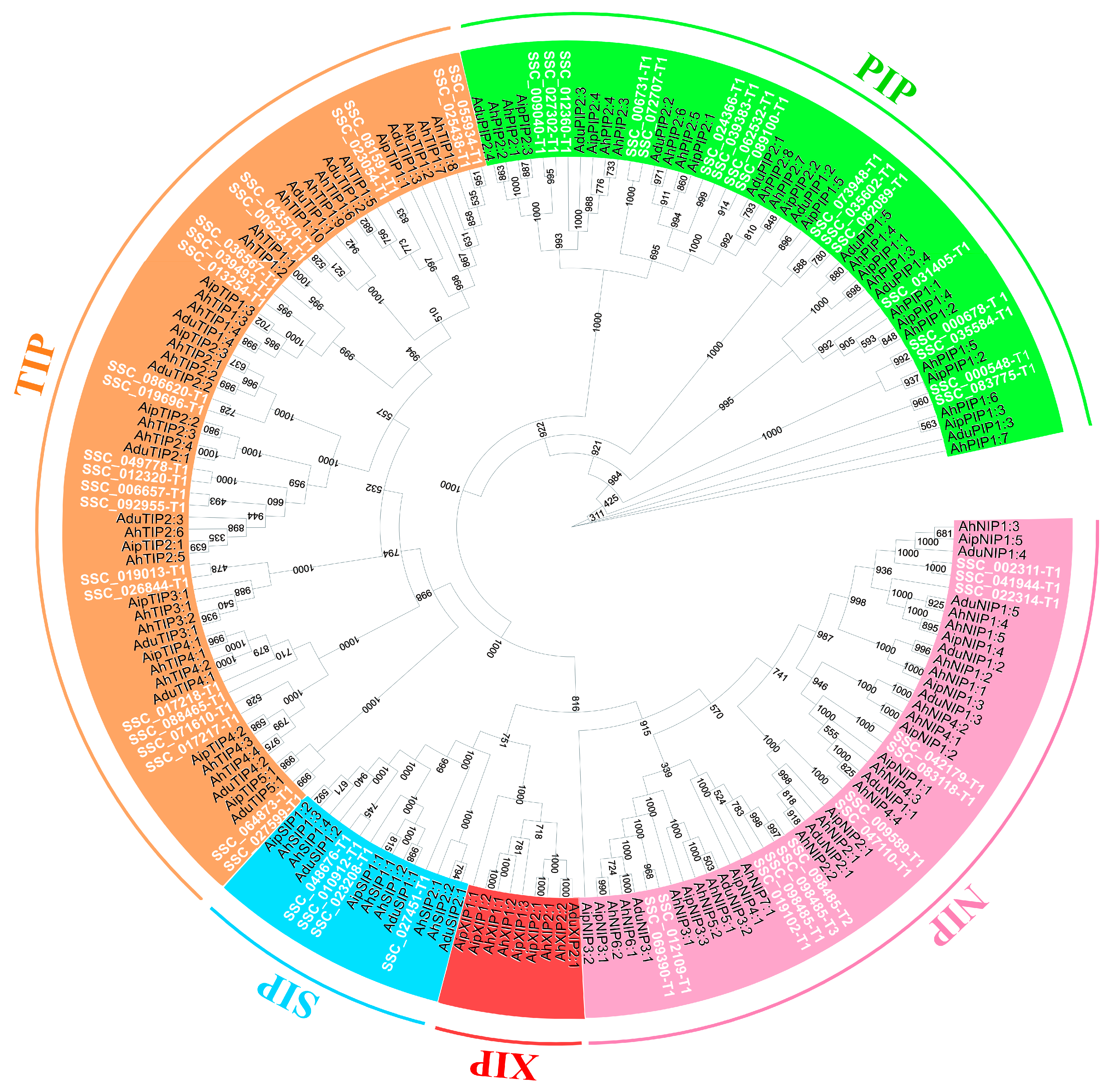
2.10. Aquaporins Mining and Identification in S. scabra Genome and Transcriptome
2.11. Identification and Annotation of Specialized Metabolite Biosynthetic Gene Clusters
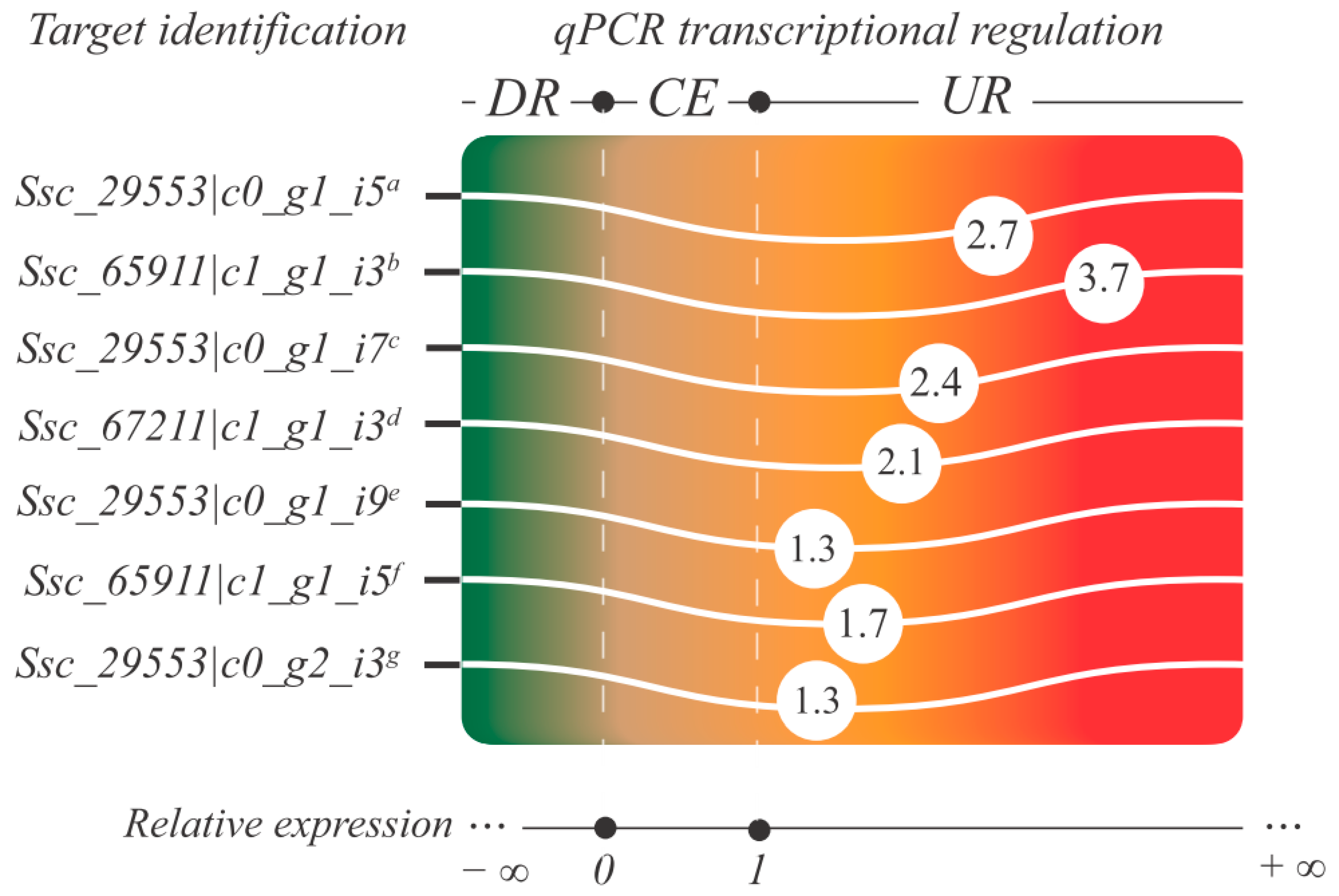
2.12. qPCR: Setup, cDNA Synthesis, Efficiency, and Relative Expression Analyses
3. Results
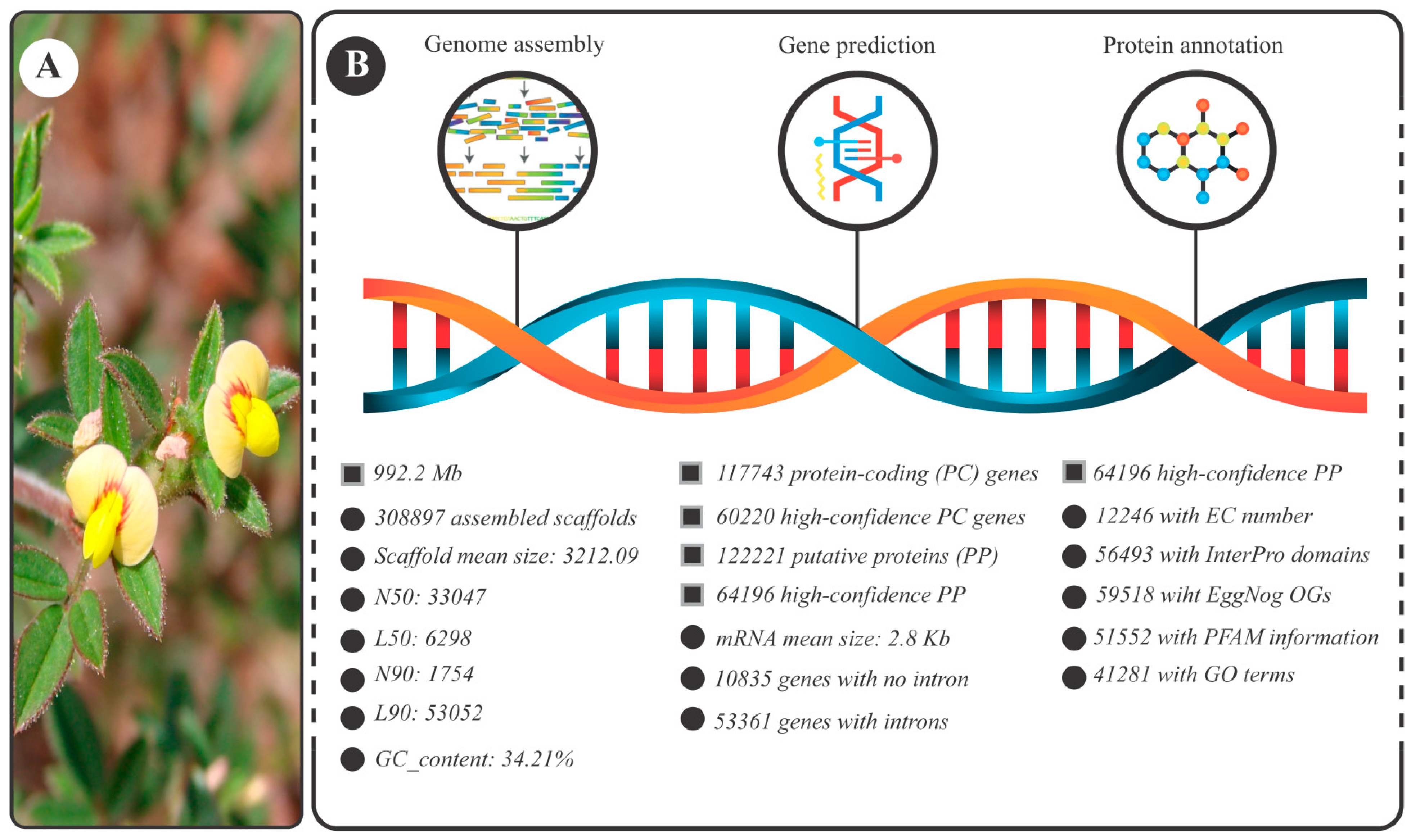
3.1. S. scabra Genome Assembly: General Data
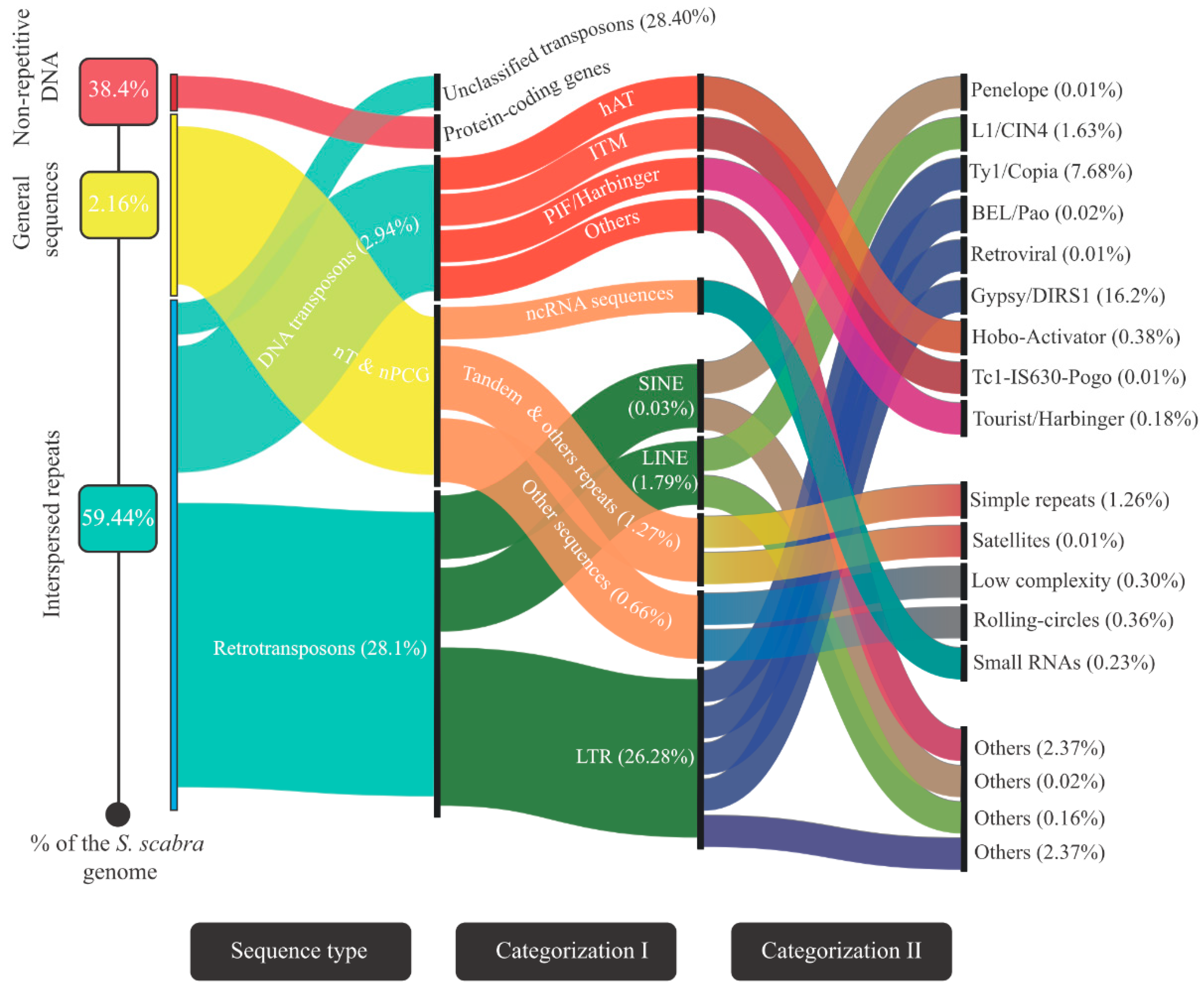
3.2. Landscape of the Stylosanthes Scabra Genome Composition
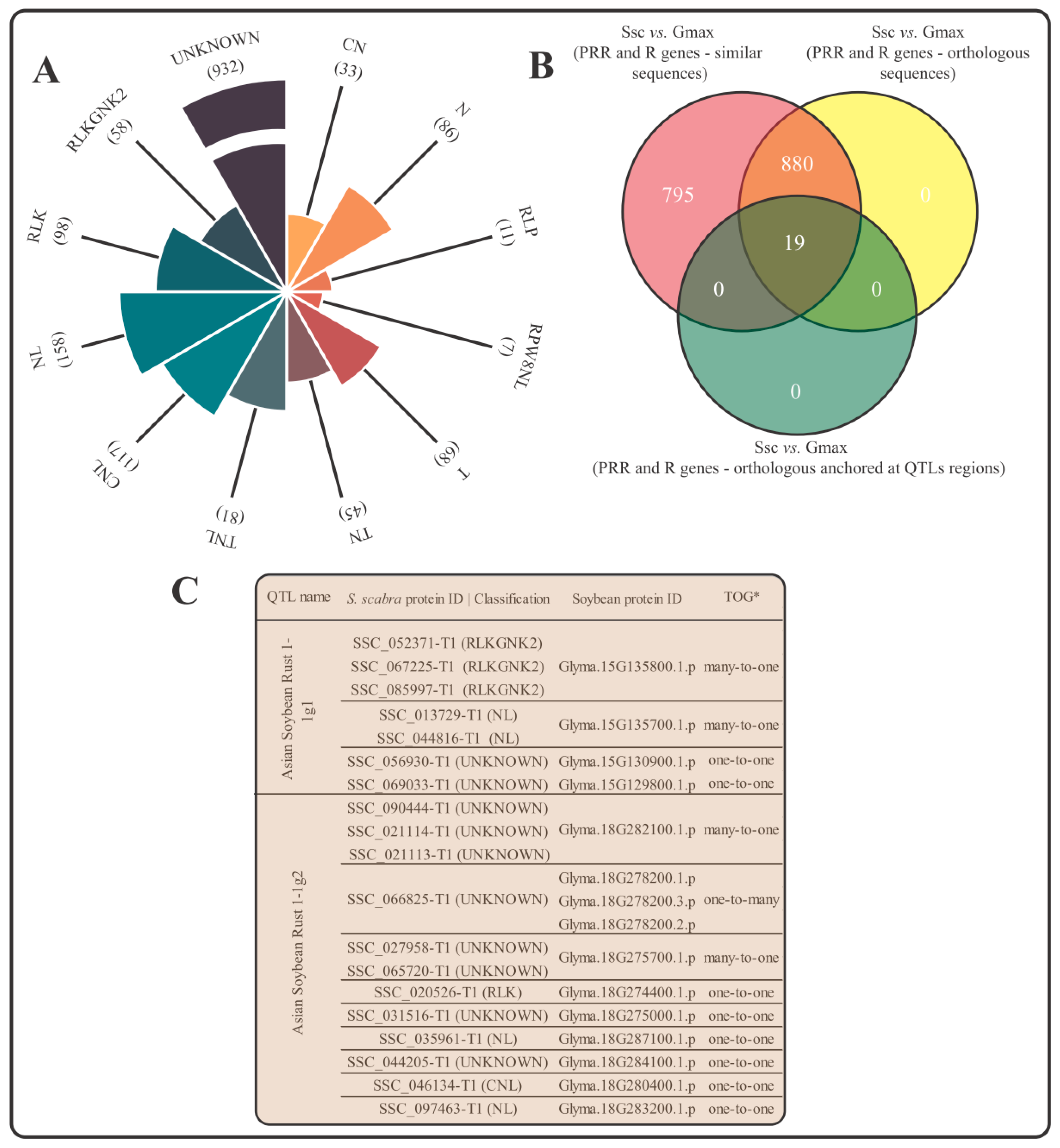
3.3. Identification of S. scabra Immune Receptors and Anchoring Analysis in Soybean QTLs Associated with Resistance to Asian Soybean Rust
3.4. Biosynthetic Gene Clusters Profile of the S. scabra Genome
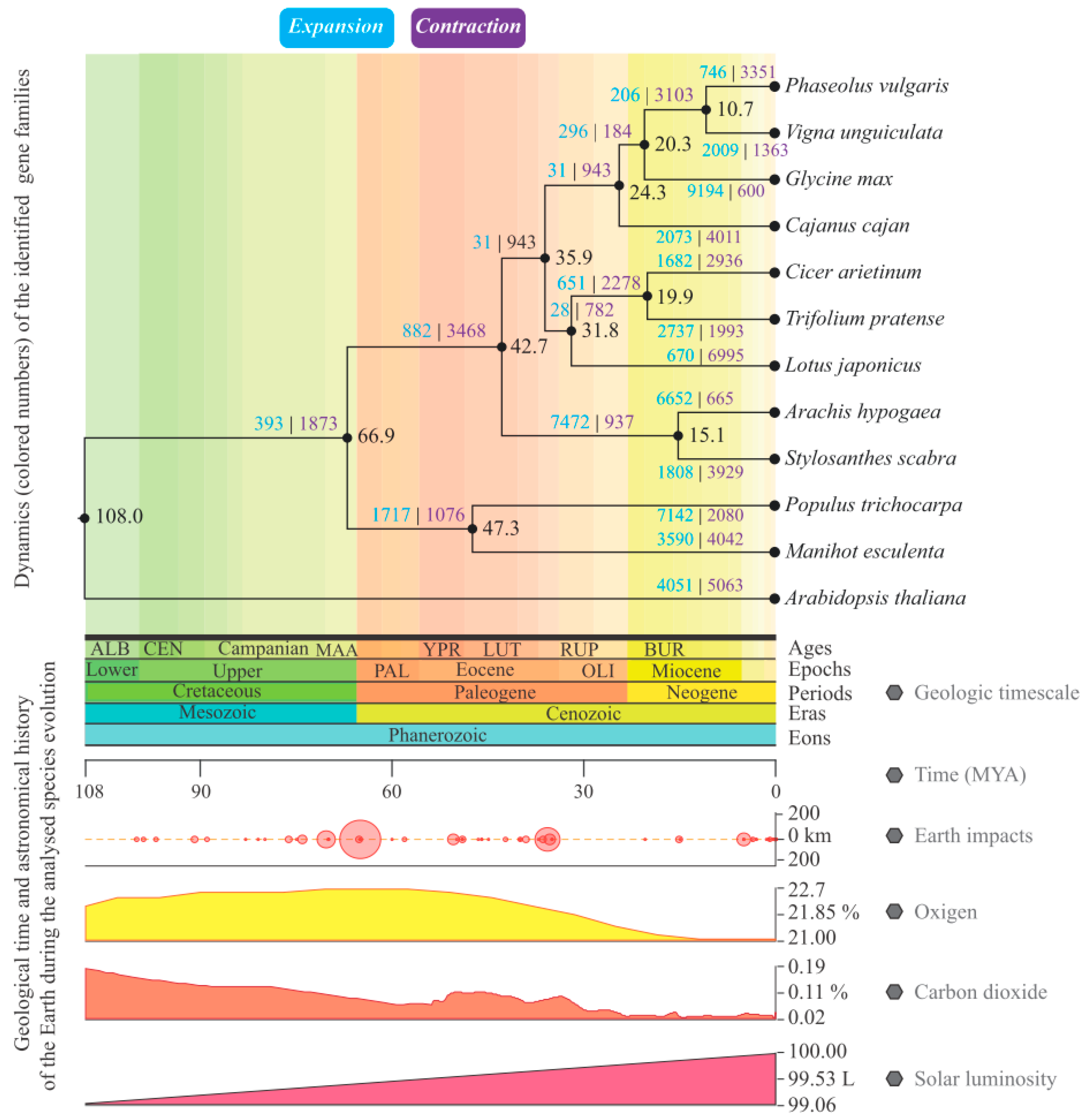
3.5. Gene Families Mining and Analysis of Their Evolutionary Dynamics
3.6. Aquaporin Gene Family: Genome-Wide Identification and Transcriptomics under Water Deficit
4. Discussion
4.1. Assembly Data and Genomic Composition
4.2. Atlas of Resistance Proteins
4.3. S. scabra as a Potential Source of Terpenes
4.4. Evaluation of Gene Families in Terms of Evolutionary Dynamics and Possible Ecophysiological Impacts
4.5. SscAQPs: Mining, Characterization, Transcriptomics, and Possible Impacts in S. scabra under Water Deficit
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Instituto Brasileiro de Geografia e Estatística (IBGE). 2014. Available online: http://www.ibge.com.br (accessed on 20 December 2022).
- Sampaio, E.V.S.B. Uso Sustentável e Conservação dos Recursos Florestais da Caatinga; Ministério do Meio Ambiente, Serviço Florestal Brasileiro: Brasília, Brazil, 2010; pp. 29–48.
- Bohnert, H.J.; Nelson, D.E.; Jensen, R.G. Adaptations to Environmental Stresses. Plant Cell 1995, 7, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, A.M.B.; Duarte, A.S.; Ometto, J.P.H.B. Modeling the potential distribution of Epiphyllum phyllanthus (L.) Haw. under future climate scenarios in the Caatinga biome. An. Acad. Bras. Ciências 2020, 92, e20180836. [Google Scholar] [CrossRef] [PubMed]
- Barcellos, A.d.O.; Ramos, A.K.B.; Vilela, L.; Junior, G.B.M. Sustentabilidade da produção animal baseada em pastagens consorciadas e no emprego de leguminosas exclusivas, na forma de banco de proteína, nos trópicos brasileiros. Rev. Bras. Zootec. 2008, 37, 51–67. [Google Scholar] [CrossRef]
- Flora do Brasil. 2022. Available online: http://florad2obrasil.jbrj.gov.br/ (accessed on 20 December 2022).
- Nagaich, D.; Tiwari, K.K.; Srivastva, N.; Chandra, A. Assessment of genetic diversity and morpho-physiological traits related to drought tolerance in Stylosanthes scabra. Acta Physiol. Plant. 2013, 35, 3127–3136. [Google Scholar] [CrossRef]
- Edye, L.A.; Topark-Ngarm, A.; Edye, L.A.; Topark-Ngarm, A. Stylosanthes Scabra Vogel. In Plant Resources of South-East Asia No. 4 Forages; Mannetje, L., Jones, R.M., Eds.; Pudoc Scientific Publishers: Wageningen, The Netherlands, 1992; pp. 219–221. Available online: http://hdl.handle.net/102.100.100/246846?index=1 (accessed on 1 April 2023).
- Chakraborty, S.; Cameron, D.F.; Irwin, J.A.G.; Edye, L.A. Quantitatively expressed resistance to anthracnose (Colletotrichum gloeosporioides) in Stylosanthes scabra. Plant Pathol. 1988, 37, 529–537. [Google Scholar] [CrossRef]
- Charchar, M.J.D.Á.; dos Anjos, J.R.N.; Gomes, A.C.; Tomaz, L.V.; Akimoto, A.K.; Karia, C.T. Avaliação de Acessos de Stylosanthes spp. em Relação à Antracnose, em Condições de Campo, no Distrito Federal, Brasil. Bol. Desenvolv. Pesqui. Embrapa 2002, 77–91. [Google Scholar]
- Marley, P.S.; Diourte, M.; Neya, A.; Rattunde, F.W. Sorghum anthracnose and sustainable management strategies in West and Central Africa. J. Sustain. Agric. 2005, 25, 43–56. [Google Scholar] [CrossRef]
- Atlas of Living Australia. 2022. Available online: https://www.ala.org.au/ (accessed on 20 December 2022).
- Ferreira-Neto, J.R.C.; de Araújo, F.C.; de Oliveira Silva, R.L.; de Melo, N.F.; Pandolfi, V.; Frosi, G.; de Lima Morais, D.A.; da Silva, M.D.; Rivas, R.; Santos, M.G.; et al. Dehydration response in Stylosanthes scabra: Transcriptional, biochemical, and physiological modulations. Physiol. Plant. 2022, 174, e13821. [Google Scholar] [CrossRef]
- Jiang, C.; Liu, L.; Li, X.; Han, R.; Wei, Y.; Yu, Y. Insights into aluminum-tolerance pathways in Stylosanthes as revealed by RNA-Seq analysis. Sci. Rep. 2018, 8, 6072. [Google Scholar] [CrossRef]
- Jiang, L.; Wu, P.; Yang, L.; Liu, C.; Guo, P.; Wang, H.; Wang, S.; Xu, F.; Zhuang, Q.; Tong, X.; et al. Transcriptomics and metabolomics reveal the induction of flavonoid biosynthesis pathway in the interaction of Stylosanthes-Colletotrichum gloeosporioides. Genomics 2021, 113, 2702–2716. [Google Scholar] [CrossRef]
- Jia, Y.; Li, X.; Liu, Q.; Hu, X.; Li, J.; Dong, R.; Liu, P.; Liu, G.; Luo, L.; Chen, Z. Physiological and transcriptomic analyses reveal the roles of secondary metabolism in the adaptive responses of Stylosanthes to manganese toxicity. BMC Genom. 2020, 21, 861. [Google Scholar] [CrossRef]
- Liu, Y.; Kong, D.; Yang, H.; Douxchamps, S.; Atieno, M.; Xu, B.; Wang, W.; Liu, G. A Transcriptomic Analysis of Stylo [Stylosanthes guianensis (Aubl.) Sw.] Provides Novel Insights into the Basis of Salinity Tolerance. Front. Sustain. Food Syst. 2022, 6, 725656. Available online: https://www.frontiersin.org/article/10.3389/fsufs.2022.725656 (accessed on 1 April 2023). [CrossRef]
- Marques, A.; Moraes, L.; Dos Santos, M.A.; Costa, I.; Costa, L.; Nunes, T.; Melo, N.; Simon, M.F.; Leitch, A.R.; Almeida, C.; et al. Origin and parental genome characterization of the allotetraploid Stylosanthes scabra Vogel (Papilionoideae, Leguminosae), an important legume pasture crop. Ann. Bot. 2018, 122, 1143–1159. [Google Scholar] [CrossRef]
- Costa, J.C.; Fracetto, G.G.M.; Fracetto, F.J.C.; Souza, T.C.; Santos, M.V.F.; Junior, M.A.L. Research Article Genetic diversity in natural populations of Stylosanthes scabra Fabaceae using ISSR markers. Genet. Mol. Res. 2019, 18, gmr18219. [Google Scholar] [CrossRef]
- Verdoucq, L.; Maurel, C. Plant aquaporins. In Advances in Botanical Research; Elsevier: Amsterdam, The Netherlands, 2018; pp. 25–56. [Google Scholar] [CrossRef]
- Maurel, C.; Boursiac, Y.; Luu, D.-T.; Santoni, V.; Shahzad, Z.; Verdoucq, L. Aquaporins in Plants. Physiol. Rev. 2015, 95, 1321–1358. [Google Scholar] [CrossRef]
- Weising, K. (Ed.) DNA Fingerprinting in Plants and Fungi; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Dolezel, J.; Göhde, W. Sex determination in dioecious plants Melandrium album and M. rubrum using high-resolution flow cytometry. Cytometry 1995, 19, 103–106. [Google Scholar] [CrossRef]
- Loureiro, J.; Rodriguez, E.; Dolezel, J.; Santos, C. Two new nuclear isolation buffers for plant DNA flow cytometry: A test with 37 species. Ann. Bot. 2007, 100, 875–888. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef]
- Nishimura, O.; Hara, Y.; Kuraku, S. gVolante for standardizing completeness assessment of genome and transcriptome assemblies. Bioinformatics 2017, 33, 3635–3637. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.K.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D.; et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [PubMed]
- Brůna, T.; Lomsadze, A.; Borodovsky, M. GeneMark-EP+: Eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom. Bioinform. 2020, 2, lqaa026. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- Drula, E.; Garron, M.-L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 2021, 36, 5516–5518. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Jin, J.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2020, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Silva, R.J.S.; Micheli, F. RRGPredictor, a set-theory-based tool for predicting pathogen-associated molecular pattern receptors (PRRs) and resistance (R) proteins from plants. Genomics 2020, 112, 2666–2676. [Google Scholar] [CrossRef]
- Shultz, J.L.; Kurunam, D.; Shopinski, K.; Iqbal, M.J.; Kazi, S.; Zobrist, K.; Bashir, R.; Yaegashi, S.; Lavu, N.; Afzal, A.J.; et al. The Soybean Genome Database (SoyGD): A browser for display of duplicated, polyploid, regions and sequence tagged sites on the integrated physical and genetic maps of Glycine max. Nucleic Acids Res. 2006, 34, D758–D765. [Google Scholar] [CrossRef]
- Shen, Y.; Gong, X. Experimental tools for the identification of specific genes in autism spectrum disorders and intellectual disability. In Neuronal and Synaptic Dysfunction in Autism Spectrum Disorder and Intellectual Disability; Elsevier: Amsterdam, The Netherlands, 2016; pp. 3–12. [Google Scholar] [CrossRef]
- Shivaraj, S.M.; Deshmukh, R.; Sonah, H.; Bélanger, R.R. Identification and characterization of aquaporin genes in Arachis duranensis and Arachis ipaensis genomes, the diploid progenitors of peanut. BMC Genom. 2019, 20, 222. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, R.; Liu, Y.; Fan, S.; Wan, S.; Zhang, X.; Li, G. The major intrinsic protein family and their function under salt-stress in peanut. Front. Genet. 2021, 12, 639585. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Kautsar, S.A.; Duran, H.G.S.; Blin, K.; Osbourn, A.; Medema, M.H. plantiSMASH: Automated identification, annotation and expression analysis of plant biosynthetic gene clusters. Nucleic Acids Res. 2017, 45, W55–W63. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef]
- Gautam, A.; Pandey, A.K. Aquaporins responses under challenging environmental conditions and abiotic stress tolerance in plants. Bot. Rev. 2021, 87, 467–495. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Bertioli, D.J.; Jenkins, J.; Clevenger, J.; Dudchenko, O.; Gao, D.; Seijo, G.; Leal-Bertioli, S.C.M.; Ren, L.; Farmer, A.D.; Pandey, M.K.; et al. The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat. Genet. 2019, 51, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Zimmermann, W.; Perovic, D.; Paterson, A.H.; Ganal, M.; Graner, A.; Stein, N. A detailed look at 7 million years of genome evolution in a 439 kb contiguous sequence at the barley Hv-eIF4E locus: Recombination, rearrangements and repeats: Seven million years of genome evolution in barley. Plant J. 2004, 41, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, Y.; Boissinot, S. On the population dynamics of junk: A review on the population genomics of transposable elements. Genes 2019, 10, 419. [Google Scholar] [CrossRef]
- Petersen, M.; Armisén, D.; Gibbs, R.A.; Hering, L.; Khila, A.; Mayer, G.; Richards, S.; Niehuis, O.; Misof, B. Diversity and evolution of the transposable element repertoire in arthropods with particular reference to insects. BMC Evol. Biol. 2019, 19, 11. [Google Scholar] [CrossRef]
- Kaul, S.; Koo, H.L.; Jenkins, J.; Rizzo, M.; Rooney, T.; Tallon, L.J.; Feldblyum, T.; Nierman, W.; Benito, M.I.; Lin, X.Y.; et al. The Arabidopsis Genome Initiative, Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef]
- Negi, P.; Rai, A.N.; Suprasanna, P. Moving through the stressed genome: Emerging regulatory roles for transposons in plant stress response. Front. Plant Sci. 2016, 7, 1448. [Google Scholar] [CrossRef]
- Deneweth, J.; Van de Peer, Y.; Vermeirssen, V. Nearby transposable elements impact plant stress gene regulatory networks: A meta-analysis in A. thaliana and S. lycopersicum. BMC Genom. 2022, 23, 18. [Google Scholar] [CrossRef]
- Munoz-Lopez, M.; Garcia-Perez, J. DNA transposons: Nature and applications in genomics. Curr. Genom. 2010, 11, 115–128. [Google Scholar] [CrossRef]
- Eickbush, T.H.; Malik, H.S. Origins and evolution of retrotransposons. In Mobile DNA II, 1st ed.; Craig, N.L., Craigie, R., Gellert, M., Lambowitz, A.M., Eds.; Wiley: Hoboken, NJ, USA, 2007; pp. 1111–1144. [Google Scholar] [CrossRef]
- Kelly, L.J.; Renny-Byfield, S.; Pellicer, J.; Macas, J.; Novák, P.; Neumann, P.; Lysak, M.A.; Day, P.D.; Berger, M.; Fay, M.F.; et al. Analysis of the giant genomes of Fritillaria (Liliaceae) indicates that a lack of DNA removal characterizes extreme expansions in genome size. New Phytol. 2015, 208, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Noman, A.; Aqeel, M.; Lou, Y. PRRs and NB-LRRs: From signal perception to activation of plant innate immunity. Int. J. Mol. Sci. 2019, 20, 1882. [Google Scholar] [CrossRef]
- Bharadwaj, R.; Kumar, S.R.; Sharma, A.; Sathishkumar, R. Plant Metabolic Gene Clusters: Evolution, Organization, and Their Applications in Synthetic Biology. Front. Plant Sci. 2021, 12, 697318. Available online: https://www.frontiersin.org/articles/10.3389/fpls.2021.697318 (accessed on 8 September 2022). [CrossRef]
- Pichersky, E.; Raguso, R.A. Why do plants produce so many terpenoid compounds? New Phytol. 2018, 220, 692–702. [Google Scholar] [CrossRef]
- Munné-Bosch, S.; Mueller, M.; Schwarz, K.; Alegre, L. Diterpenes and antioxidative protection in drought-stressed Salvia officinalis plants. J. Plant Physiol. 2001, 158, 1431–1437. [Google Scholar] [CrossRef]
- Houston, K.; Tucker, M.R.; Chowdhury, J.; Shirley, N.; Little, A. The Plant Cell Wall: A Complex and Dynamic Structure as Revealed by the Responses of Genes under Stress Conditions. Front. Plant Sci. 2016, 7, 984. Available online: https://www.frontiersin.org/articles/10.3389/fpls.2016.00984 (accessed on 8 September 2022). [CrossRef]
- Tenhaken, R. Cell Wall Remodeling under Abiotic Stress. Front. Plant Sci. 2015, 5, 771. Available online: https://www.frontiersin.org/articles/10.3389/fpls.2014.00771 (accessed on 1 April 2023). [CrossRef]
- Gall, H.L.; Philippe, F.; Domon, J.-M.; Gillet, F.; Pelloux, J.; Rayon, C. Cell wall metabolism in response to abiotic stress. Plants 2015, 4, 112–166. [Google Scholar] [CrossRef]
- Demidchik, V. Mechanisms of oxidative stress in plants: From classical chemistry to cell biology. Environ. Exp. Bot. 2015, 109, 212–228. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Bhuyan, M.H.M.B.; Zulfiqar, F.; Raza, A.; Mohsin, S.M.; Mahmud, J.A.; Fujita, M.; Fotopoulos, V. Reactive oxygen species and antioxidant defense in plants under abiotic stress: Revisiting the crucial role of a universal defense regulator. Antioxidants 2020, 9, 681. [Google Scholar] [CrossRef]
- Rohman, M.; Islam, R.; Monsur, M.B.; Amiruzzaman, M.; Fujita, M.; Hasanuzzaman, M. Trehalose protects maize plants from salt stress and phosphorus deficiency. Plants 2019, 8, 568. [Google Scholar] [CrossRef]
- Joshi, R.; Sahoo, K.K.; Singh, A.K.; Anwar, K.; Pundir, P.; Gautam, R.K.; Krishnamurthy, S.L.; Sopory, S.K.; Pareek, A.; Singla-Pareek, S.L. Enhancing trehalose biosynthesis improves yield potential in marker-free transgenic rice under drought, saline, and sodic conditions. J. Exp. Bot. 2020, 71, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Hayat, S.; Hayat, Q.; Alyemeni, M.N.; Wani, A.S.; Pichtel, J.; Ahmad, A. Role of proline under changing environments: A review. Plant Signal Behav. 2012, 7, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, U.K.; Islam, M.N.; Siddiqui, M.N.; Cao, X.; Khan, M.A.R. Proline, a multifaceted signalling molecule in plant responses to abiotic stress: Understanding the physiological mechanisms. Plant Biol. 2022, 24, 227–239. [Google Scholar] [CrossRef]
- Cardoso, D.; Pennington, R.T.; de Queiroz, L.P.; Boatwright, J.S.; Van Wyk, B.-E.; Wojciechowski, M.F.; Lavin, M. Reconstructing the deep-branching relationships of the papilionoid legumes. S. Afr. J. Bot. 2013, 89, 58–75. [Google Scholar] [CrossRef]
- Franco, A.L.; Figueredo, A.; Pereira, L.d.M.; de Sousa, S.M.; Souza, G.; Carvalho, M.A.; Simon, M.F.; Viccini, L.F. Low cytomolecular diversification in the genus Stylosanthes Sw. (Papilionoideae, Leguminosae). Genet. Mol. Biol. 2020, 43, e20180250. [Google Scholar] [CrossRef]
- Feng, Z.-J.; Liu, N.; Zhang, G.-W.; Niu, F.-G.; Xu, S.-C.; Gong, Y.-M. Investigation of the AQP family in soybean and the promoter activity of TIP2;6 in heat stress and hormone responses. Int. J. Mol. Sci. 2019, 20, 262. [Google Scholar] [CrossRef]
- Shang, X.; Cao, Y.; Ma, L. Alternative splicing in plant genes: A means of regulating the environmental fitness of plants. Int. J. Mol. Sci. 2017, 18, 432. [Google Scholar] [CrossRef]
- Javot, H.; Lauvergeat, V.; Santoni, V.; Martin-Laurent, F.; Güçlü, J.; Vinh, J.; Heyes, J.; Franck, K.I.; Schäffner, A.R.; Bouchez, D.; et al. Role of a single aquaporin isoform in root water uptake. Plant Cell 2003, 15, 509–522. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Pickel, S.U.; Jennings, J.; Törnroth-Horsefield, S.; Conner, M.T.; Bill, R.M.; Conner, A.C. Water channel pore size determines exclusion properties but not solute selectivity. Sci. Rep. 2019, 9, 20369. [Google Scholar] [CrossRef]
- Savage, D.F.; Egea, P.F.; Robles-Colmenares, Y.; O’Connell, J.D., III; Stroud, R.M. Architecture and selectivity in aquaporins: 2.5 Å X-Ray structure of aquaporin Z. PLoS Biol. 2003, 1, e72. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira-Neto, J.R.C.; da Silva, M.D.; Binneck, E.; de Melo, N.F.; da Silva, R.H.; de Melo, A.L.T.M.; Pandolfi, V.; Bustamante, F.d.O.; Brasileiro-Vidal, A.C.; Benko-Iseppon, A.M. Bridging the Gap: Combining Genomics and Transcriptomics Approaches to Understand Stylosanthes scabra, an Orphan Legume from the Brazilian Caatinga. Plants 2023, 12, 3246. https://doi.org/10.3390/plants12183246
Ferreira-Neto JRC, da Silva MD, Binneck E, de Melo NF, da Silva RH, de Melo ALTM, Pandolfi V, Bustamante FdO, Brasileiro-Vidal AC, Benko-Iseppon AM. Bridging the Gap: Combining Genomics and Transcriptomics Approaches to Understand Stylosanthes scabra, an Orphan Legume from the Brazilian Caatinga. Plants. 2023; 12(18):3246. https://doi.org/10.3390/plants12183246
Chicago/Turabian StyleFerreira-Neto, José Ribamar Costa, Manassés Daniel da Silva, Eliseu Binneck, Natoniel Franklin de Melo, Rahisa Helena da Silva, Ana Luiza Trajano Mangueira de Melo, Valesca Pandolfi, Fernanda de Oliveira Bustamante, Ana Christina Brasileiro-Vidal, and Ana Maria Benko-Iseppon. 2023. "Bridging the Gap: Combining Genomics and Transcriptomics Approaches to Understand Stylosanthes scabra, an Orphan Legume from the Brazilian Caatinga" Plants 12, no. 18: 3246. https://doi.org/10.3390/plants12183246
APA StyleFerreira-Neto, J. R. C., da Silva, M. D., Binneck, E., de Melo, N. F., da Silva, R. H., de Melo, A. L. T. M., Pandolfi, V., Bustamante, F. d. O., Brasileiro-Vidal, A. C., & Benko-Iseppon, A. M. (2023). Bridging the Gap: Combining Genomics and Transcriptomics Approaches to Understand Stylosanthes scabra, an Orphan Legume from the Brazilian Caatinga. Plants, 12(18), 3246. https://doi.org/10.3390/plants12183246