
Genome-Wide Association Studies Using 3VmrMLM Model Provide New Insights into Branched-Chain Amino Acid Contents in Rice Grains

Abstract
:1. Introduction
2. Results
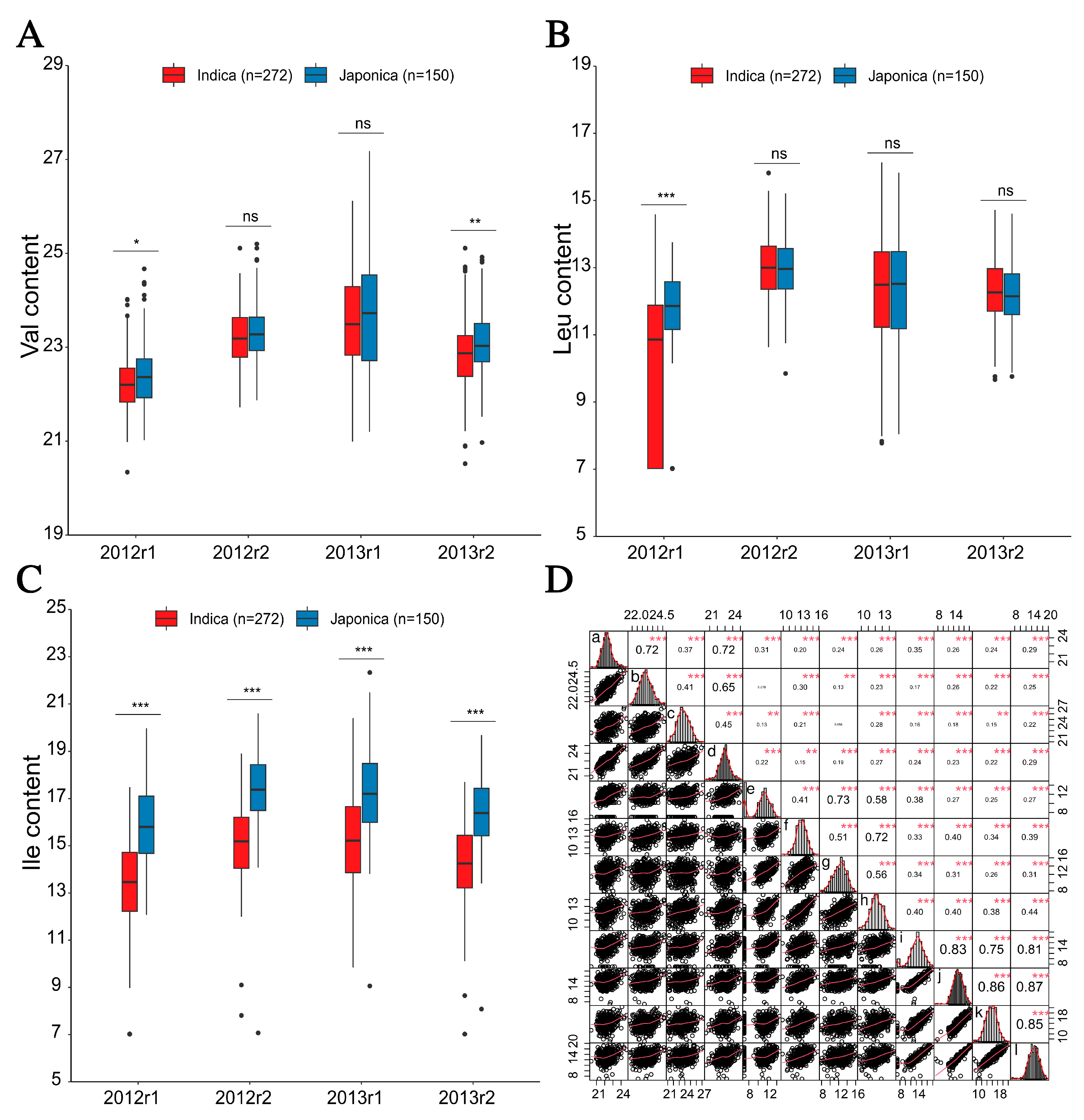
2.1. Phenotypic Variation and Correlation Analysis
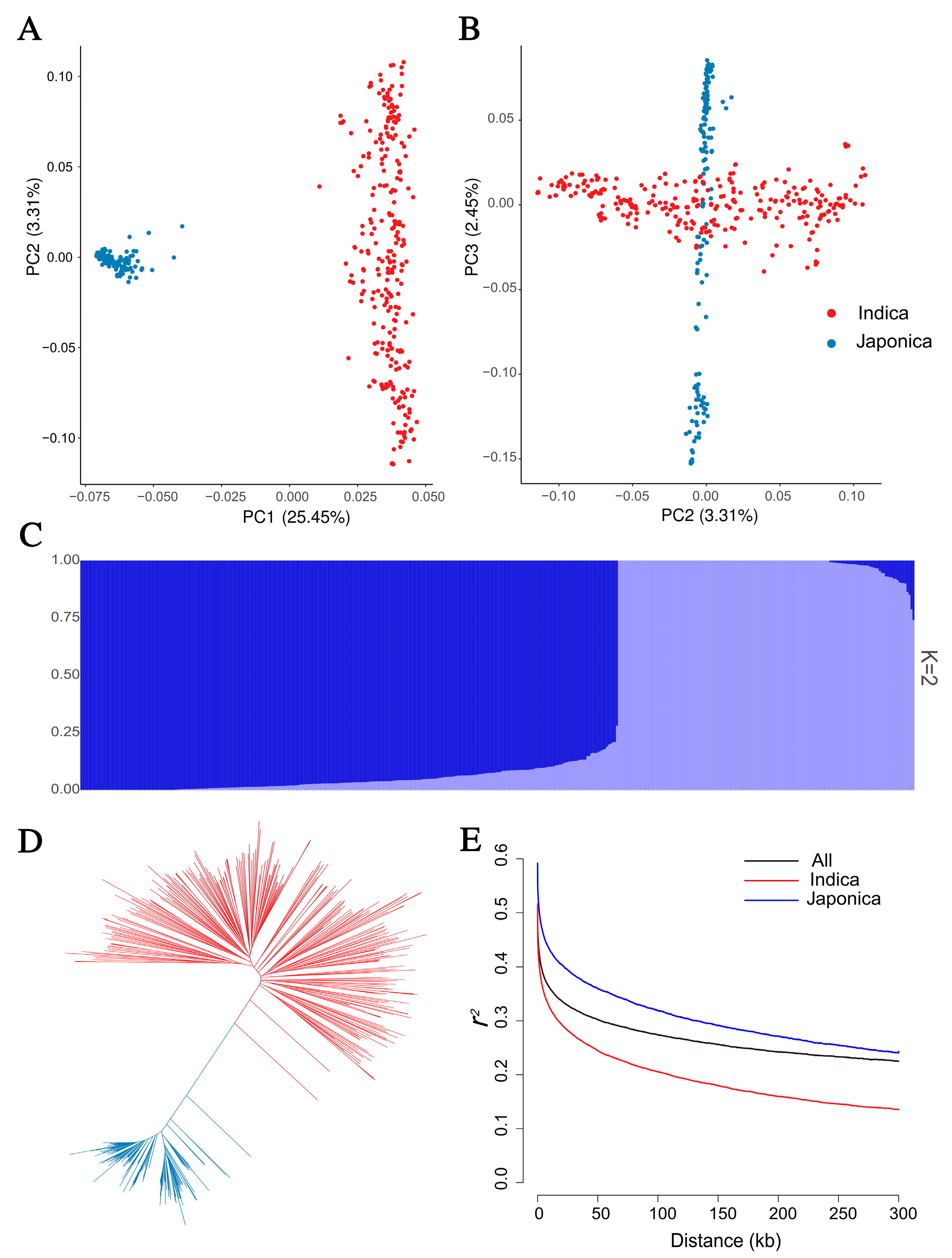
2.2. Population Analysis of 422 Rice Accessions
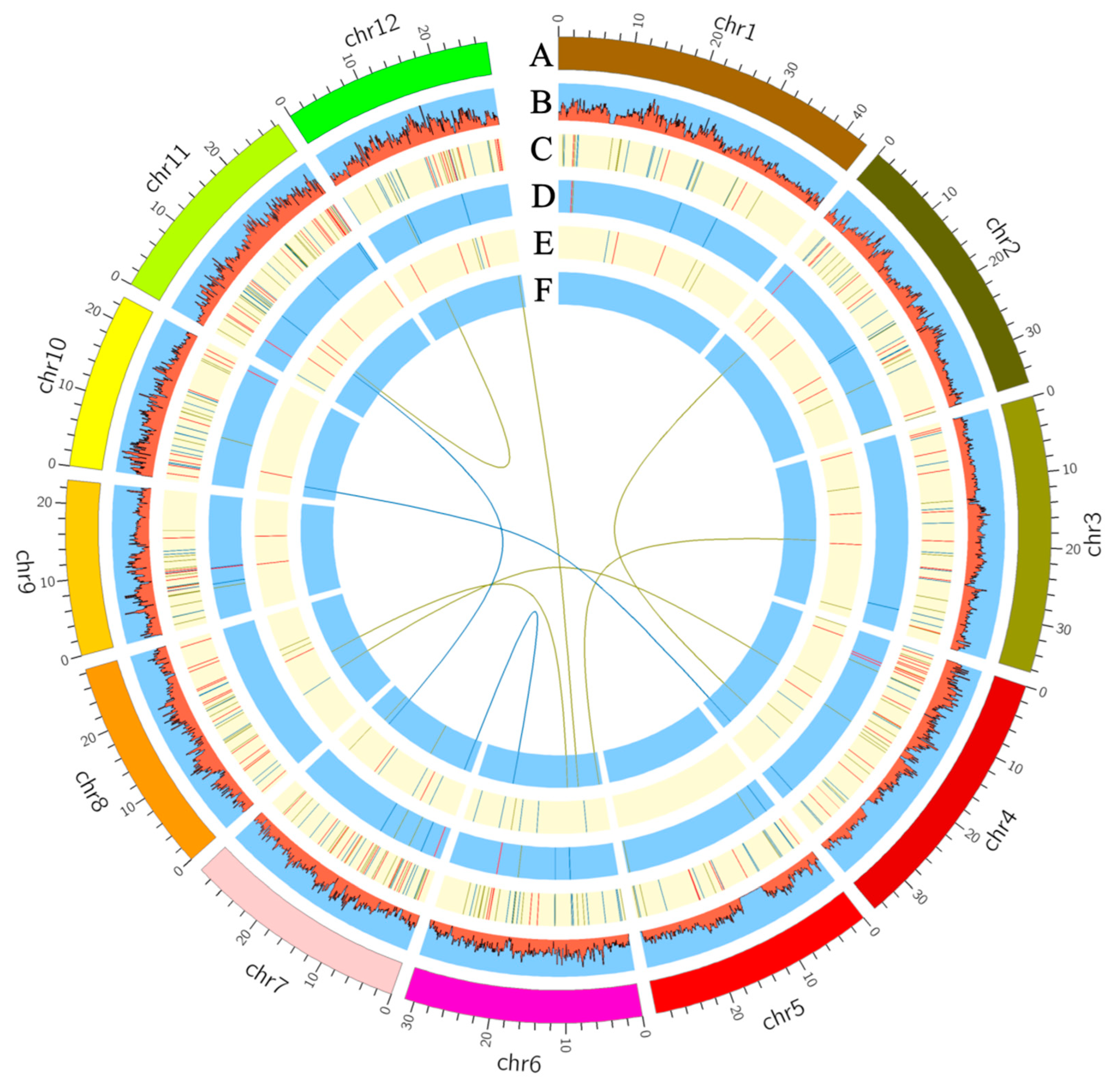
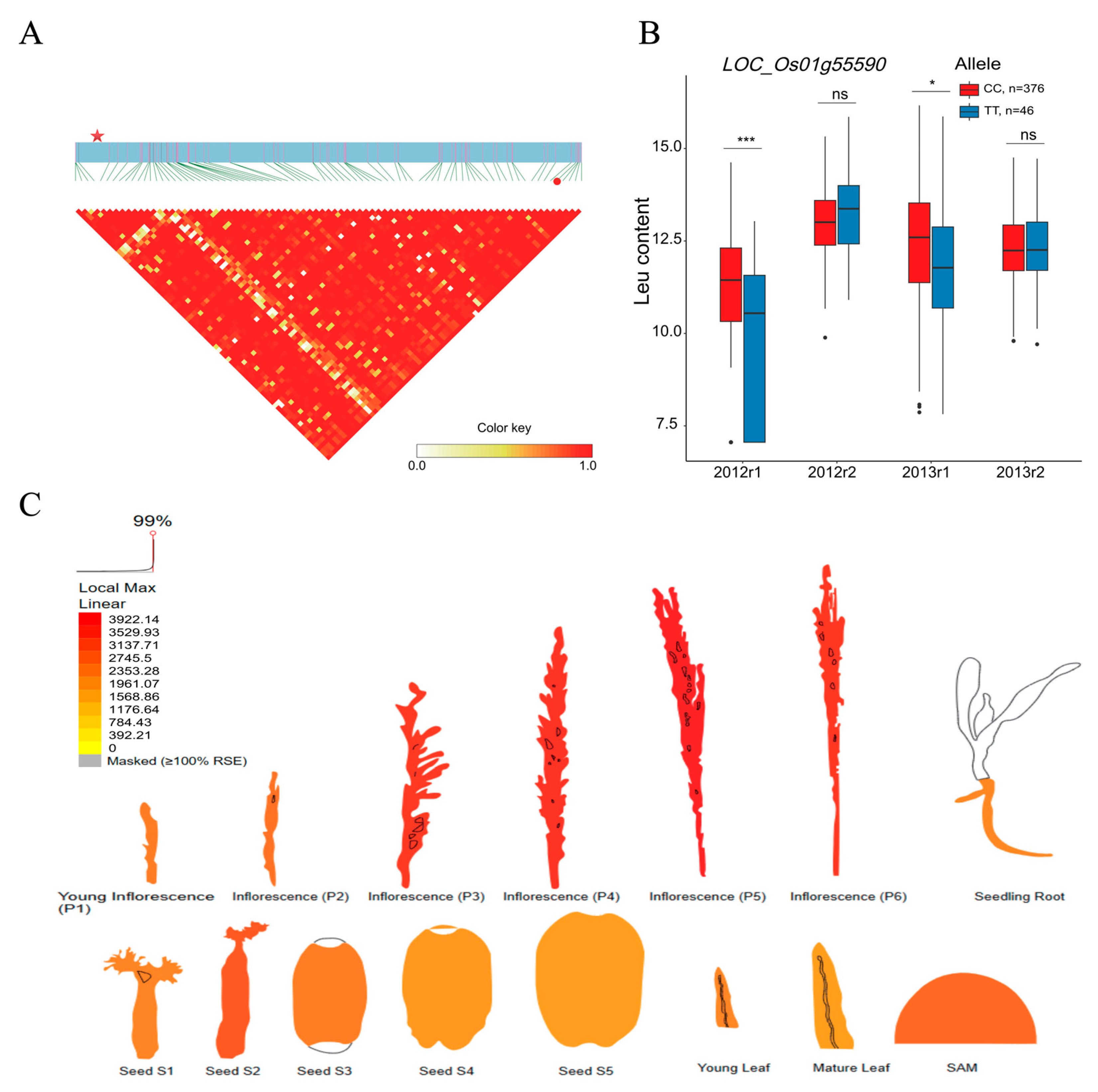
2.3. BCAA-Level-Associated QTNs and Candidate Genes
2.4. BCAA-Level-Related QEIs and Candidate Genes
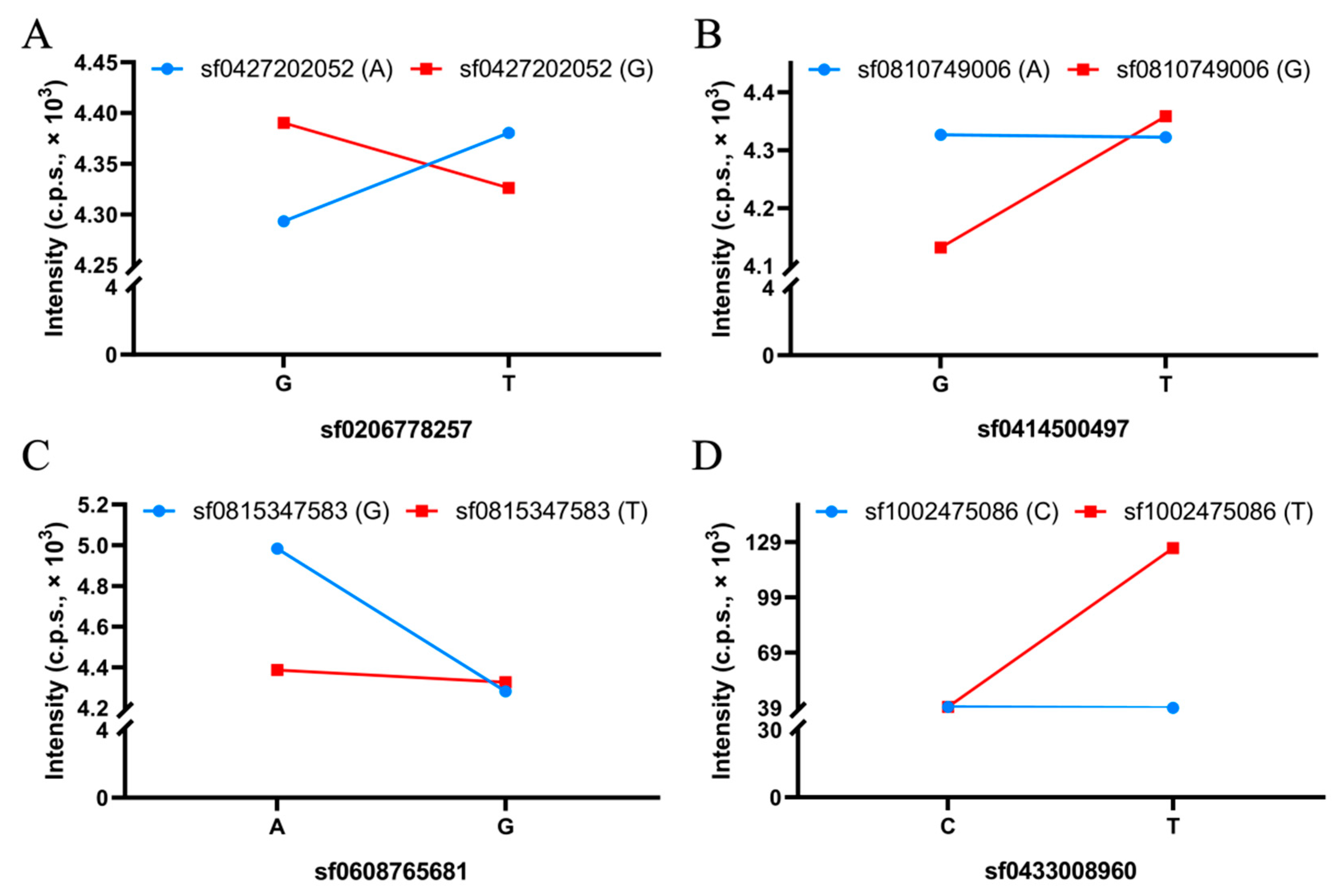
2.5. BCAA-Level-Related QQIs
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Sequencing
4.2. Metabolite Profiling
4.3. Population Stratification and Linkage Disequilibrium Analysis
4.4. Genome-Wide Association Study
4.5. Candidate Gene Prediction and Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ufaz, S.; Galili, G. Improving the content of essential amino acids in crop plants: Goals and opportunities. Plant Physiol. 2008, 147, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Fukagawa, N.K.; Ziska, L.H. Rice: Importance for Global Nutrition. J. Nutr. Sci. Vitaminol. 2019, 65, S2–S3. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; An, G.; Weber, A.P.M.; Zhang, D. Prospects for rice in 2050. Plant Cell Environ. 2023, 46, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Fang, C.; Zhang, Y.; Fernie, A.R.; Luo, J. Plant metabolism paves the way for breeding crops with high nutritional value and stable yield. Sci. China Life Sci. 2021, 64, 2202–2205. [Google Scholar] [CrossRef] [PubMed]
- Neinast, M.; Murashige, D.; Arany, Z. Branched Chain Amino Acids. Annu. Rev. Physiol. 2019, 81, 139–164. [Google Scholar] [CrossRef] [PubMed]
- Galili, G.; Amir, R.; Fernie, A.R. The Regulation of Essential Amino Acid Synthesis and Accumulation in Plants. Annu. Rev. Plant Biol. 2016, 67, 153–178. [Google Scholar] [CrossRef] [PubMed]
- Le Couteur, D.G.; Solon-Biet, S.M.; Cogger, V.C.; Ribeiro, R.; de Cabo, R.; Raubenheimer, D.; Cooney, G.J.; Simpson, S.J. Branched chain amino acids, aging and age-related health. Ageing Res. Rev. 2020, 64, 101198. [Google Scholar] [CrossRef]
- McGarrah, R.W.; White, P.J. Branched-chain amino acids in cardiovascular disease. Nat. Rev. Cardiol. 2023, 20, 77–89. [Google Scholar] [CrossRef]
- Wolfe, R.R. Branched-chain amino acids and muscle protein synthesis in humans: Myth or reality? J. Int. Soc. Sports Nutr. 2017, 14, 30. [Google Scholar] [CrossRef]
- Bifari, F.; Ruocco, C.; Decimo, I.; Fumagalli, G.; Valerio, A.; Nisoli, E. Amino acid supplements and metabolic health: A potential interplay between intestinal microbiota and systems control. Genes Nutr. 2017, 12, 27. [Google Scholar] [CrossRef]
- Tibbs Cortes, L.; Zhang, Z.; Yu, J. Status and prospects of genome-wide association studies in plants. Plant Genome 2021, 14, e20077. [Google Scholar] [CrossRef] [PubMed]
- Demirjian, C.; Vailleau, F.; Berthome, R.; Roux, F. Genome-wide association studies in plant pathosystems: Success or failure? Trends Plant Sci. 2023, 28, 471–485. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Gai, J. Genome-Wide Association Studies (GWAS). Methods Mol. Biol. 2023, 2638, 123–146. [Google Scholar] [PubMed]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Jia, Z.; Dunwell, J.M. Editorial: The Applications of New Multi-Locus GWAS Methodologies in the Genetic Dissection of Complex Traits. Front. Plant Sci. 2019, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Segura, V.; Vilhjalmsson, B.J.; Platt, A.; Korte, A.; Seren, U.; Long, Q.; Nordborg, M. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 2012, 44, 825–830. [Google Scholar] [CrossRef]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Wang, S.B.; Feng, J.Y.; Ren, W.L.; Huang, B.; Zhou, L.; Wen, Y.J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef]
- Ren, W.; Wen, Y.; Dunwell, J.; Zhang, Y. pKWmEB: Integration of Kruskal-Wallis test with empirical Bayes under polygenic background control for multi-locus genome-wide association study. Heredity 2018, 120, 208–218. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, J.; Ni, Y.; Wen, Y.; Niu, Y.; Tamba, C.; Yue, C.; Song, Q.; Zhang, Y. pLARmEB: Integration of least angle regression with empirical Bayes for multilocus genome-wide association studies. Heredity 2017, 118, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Zhang, H.; Ni, Y.; Huang, B.; Zhang, J.; Feng, J.; Wang, S.; Dunwell, J.; Zhang, Y.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef]
- Tamba, C.; Ni, Y.; Zhang, Y. Iterative sure independence screening EM-Bayesian LASSO algorithm for multi-locus genome-wide association studies. PLoS Comput. Biol. 2017, 13, e1005357. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Y.W.; Zhang, Z.C.; Xiang, Y.; Liu, M.H.; Zhou, Y.H.; Zuo, J.F.; Zhang, H.Q.; Chen, Y.; Zhang, Y.M. A compressed variance component mixed model for detecting QTNs, and QTN-by-environment and QTN-by-QTN interactions in genome-wide association studies. Mol. Plant 2022, 15, 630–650. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. Metabolite-based genome-wide association studies in plants. Curr. Opin. Plant Biol. 2015, 24, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Luo, J. Metabolic GWAS-based dissection of genetic bases underlying the diversity of plant metabolism. Plant J. 2019, 97, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Luo, J.; Wang, S. The Diversity of Nutritional Metabolites: Origin, Dissection, and Application in Crop Breeding. Front. Plant Sci. 2019, 10, 1028. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; He, L.; Yang, J.; Fernie, A.R.; Luo, J. Natural variance at the interface of plant primary and specialized metabolism. Curr. Opin. Plant Biol. 2022, 67, 102201. [Google Scholar] [CrossRef]
- Sun, Y.; Shi, Y.; Liu, G.; Yao, F.; Zhang, Y.; Yang, C.; Guo, H.; Liu, X.; Jin, C.; Luo, J. Natural variation in the OsbZIP18 promoter contributes to branched-chain amino acid levels in rice. New Phytol. 2020, 228, 1548–1558. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, Y.; Sun, Y.; Xie, Z.; Luo, Y.; Long, Q.; Feng, J.; Liu, X.; Wang, B.; He, D.; et al. Natural variations of OsAUX5, a target gene of OsWRKY78, control the contents of neutral essential amino acids in rice grains. Mol. Plant 2022, 16, 322–336. [Google Scholar] [CrossRef]
- Chen, W.; Gao, Y.; Xie, W.; Gong, L.; Lu, K.; Wang, W.; Li, Y.; Liu, X.; Zhang, H.; Dong, H.; et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 2014, 46, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, W.; Peng, M.; Gong, L.; Gao, Y.; Wan, J.; Wang, S.; Shi, L.; Zhou, B.; Li, Z.; et al. Comparative and parallel genome-wide association studies for metabolic and agronomic traits in cereals. Nat. Commun. 2016, 7, 12767. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, S.; Huang, Z.; Zhang, S.; Liao, Q.; Zhang, C.; Lin, T.; Qin, M.; Peng, M.; Yang, C.; et al. Rewiring of the fruit metabolome in tomato breeding. Cell 2018, 172, 249–261.e212. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Duan, H.; Sun, Y.; Hu, R.; Wu, B.; Lin, J.; Deng, W.; Li, Y.; Zheng, H. Genome-Wide Association Study With Growth-Related Traits and Secondary Metabolite Contents in Red- and White-Heart Chinese Fir. Front. Plant Sci. 2022, 13, 922007. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wang, H.; Sui, Y.; Miao, Y.; Jin, C.; Luo, J. Genome-wide association studies of five free amino acid levels in rice. Front. Plant Sci. 2022, 13, 1048860. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Tsai, A.; Dattmore, D.A.; Stives, D.P.; Chitrakar, I.; D’Alessandro, A.M.; Patil, S.; Hicks, K.A.; French, J.B. Crystal structure of E. coli PRPP synthetase. BMC Struct. Biol. 2019, 19, 1. [Google Scholar] [CrossRef] [PubMed]
- Hove-Jensen, B.; Andersen, K.R.; Kilstrup, M.; Martinussen, J.; Switzer, R.L.; Willemoes, M. Phosphoribosyl Diphosphate (PRPP): Biosynthesis, Enzymology, Utilization, and Metabolic Significance. Microbiol. Mol. Biol. Rev. 2017, 81, e00040-16. [Google Scholar] [CrossRef]
- Zrenner, R.; Stitt, M.; Sonnewald, U.; Boldt, R. Pyrimidine and purine biosynthesis and degradation in plants. Annu. Rev. Plant Biol. 2006, 57, 805–836. [Google Scholar] [CrossRef]
- Witte, C.P.; Herde, M. Nucleotide Metabolism in Plants. Plant Physiol. 2020, 182, 63–78. [Google Scholar] [CrossRef]
- Yang, Q.; Zhao, D.; Liu, Q. Connections Between Amino Acid Metabolisms in Plants: Lysine as an Example. Front. Plant Sci. 2020, 11, 928. [Google Scholar] [CrossRef]
- Gaufichon, L.; Reisdorf-Cren, M.; Rothstein, S.J.; Chardon, F.; Suzuki, A. Biological functions of asparagine synthetase in plants. Plant Sci. 2010, 179, 141–153. [Google Scholar] [CrossRef]
- Reitzer, L. Biosynthesis of Glutamate, Aspartate, Asparagine, L-Alanine, and D-Alanine. EcoSal Plus 2004, 1, 10–1128. [Google Scholar] [CrossRef]
- Azevedo, R.A.; Lancien, M.; Lea, P.J. The aspartic acid metabolic pathway, an exciting and essential pathway in plants. Amino Acids 2006, 30, 143–162. [Google Scholar] [CrossRef] [PubMed]
- Binder, S. Branched-Chain Amino Acid Metabolism in Arabidopsis thaliana. Arab. Book 2010, 8, e0137. [Google Scholar] [CrossRef] [PubMed]
- Maloney, G.S.; Kochevenko, A.; Tieman, D.M.; Tohge, T.; Krieger, U.; Zamir, D.; Taylor, M.G.; Fernie, A.R.; Klee, H.J. Characterization of the branched-chain amino acid aminotransferase enzyme family in tomato. Plant Physiol. 2010, 153, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Angelovici, R.; Lipka, A.E.; Deason, N.; Gonzalez-Jorge, S.; Lin, H.; Cepela, J.; Buell, R.; Gore, M.A.; Dellapenna, D. Genome-wide analysis of branched-chain amino acid levels in Arabidopsis seeds. Plant Cell 2013, 25, 4827–4843. [Google Scholar] [CrossRef] [PubMed]
- Binder, S.; Knill, T.; Schuster, J. Branched-chain amino acid metabolism in higher plants. Physiol. Plant 2007, 129, 68–78. [Google Scholar] [CrossRef]
- Campbell, M.A.; Patel, J.K.; Meyers, J.L.; Myrick, L.C.; Gustin, J.L. Genes encoding for branched-chain amino acid aminotransferase are differentially expressed in plants. Plant Physiol. Biochem. 2001, 39, 855–860. [Google Scholar] [CrossRef]
- Jin, C.; Sun, Y.; Shi, Y.; Zhang, Y.; Chen, K.; Li, Y.; Liu, G.; Yao, F.; Cheng, D.; Li, J.; et al. Branched-chain amino acids regulate plant growth by affecting the homeostasis of mineral elements in rice. Sci. China Life Sci. 2019, 62, 1107–1110. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Lopez-Maside, L.; Donapetry-Garcia, C.; Fernandez-Fernandez, C.; Sixto-Leal, C. Enzymes involved in branched-chain amino acid metabolism in humans. Amino Acids 2017, 49, 1005–1028. [Google Scholar] [CrossRef]
- Bowman, C.E.; Wolfgang, M.J. Role of the malonyl-CoA synthetase ACSF3 in mitochondrial metabolism. Adv. Biol. Regul. 2019, 71, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kim, H.U.; Weng, H.; Browse, J. Malonyl-CoA synthetase, encoded by ACYL ACTIVATING ENZYME13, is essential for growth and development of Arabidopsis. Plant Cell 2011, 23, 2247–2262. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, H.; Yu, O. A plant malonyl-CoA synthetase enhances lipid content and polyketide yield in yeast cells. Appl. Microbiol. Biotechnol. 2014, 98, 5435–5447. [Google Scholar] [CrossRef] [PubMed]
- Igamberdiev, A.U.; Kleczkowski, L.A. The Glycerate and Phosphorylated Pathways of Serine Synthesis in Plants: The Branches of Plant Glycolysis Linking Carbon and Nitrogen Metabolism. Front. Plant Sci. 2018, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Ros, R.; Munoz-Bertomeu, J.; Krueger, S. Serine in plants: Biosynthesis, metabolism, and functions. Trends Plant Sci. 2014, 19, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, T.M.; Nunes Nesi, A.; Araujo, W.L.; Braun, H.P. Amino Acid Catabolism in Plants. Mol. Plant 2015, 8, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Li, Y.; Xiong, L.; Yan, T.; Zou, J.; Dai, Z.; Tang, G.; Sun, K.; Luan, X.; Yang, W.; et al. Development of Wide-Compatible Indica Lines by Pyramiding Multiple Neutral Alleles of Indica-Japonica Hybrid Sterility Loci. Front. Plant Sci. 2022, 13, 890568. [Google Scholar] [CrossRef]
- Tamisier, L.; Szadkowski, M.; Nemouchi, G.; Lefebvre, V.; Szadkowski, E.; Duboscq, R.; Santoni, S.; Sarah, G.; Sauvage, C.; Palloix, A.; et al. Genome-wide association mapping of QTLs implied in potato virus Y population sizes in pepper: Evidence for widespread resistance QTL pyramiding. Mol. Plant Pathol. 2020, 21, 3–16. [Google Scholar] [CrossRef]
- Ashikari, M.; Matsuoka, M. Identification, isolation and pyramiding of quantitative trait loci for rice breeding. Trends Plant Sci. 2006, 11, 344–350. [Google Scholar] [CrossRef]
- Cruppe, G.; Lemes da Silva, C.; Lollato, R.; Fritz, A.; Kuhnem, P.; Cruz, C.D.; Calderon, L.; Valent, B. QTL pyramiding provides marginal improvement in 2NvS-based wheat blast resistance. Plant Dis. 2023. [Google Scholar] [CrossRef]
- Pilet-Nayel, M.L.; Moury, B.; Caffier, V.; Montarry, J.; Kerlan, M.C.; Fournet, S.; Durel, C.E.; Delourme, R. Quantitative Resistance to Plant Pathogens in Pyramiding Strategies for Durable Crop Protection. Front. Plant Sci. 2017, 8, 1838. [Google Scholar] [CrossRef] [PubMed]
- Crossa, J.; Perez-Rodriguez, P.; Cuevas, J.; Montesinos-Lopez, O.; Jarquin, D.; de Los Campos, G.; Burgueno, J.; Gonzalez-Camacho, J.M.; Perez-Elizalde, S.; Beyene, Y.; et al. Genomic Selection in Plant Breeding: Methods, Models, and Perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Newell, M.A.; Jannink, J.L. Genomic selection in plant breeding. Methods Mol. Biol. 2014, 1145, 117–130. [Google Scholar] [PubMed]
- Bhat, J.A.; Ali, S.; Salgotra, R.K.; Mir, Z.A.; Dutta, S.; Jadon, V.; Tyagi, A.; Mushtaq, M.; Jain, N.; Singh, P.K.; et al. Genomic Selection in the Era of Next Generation Sequencing for Complex Traits in Plant Breeding. Front. Genet. 2016, 7, 221. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, X.; Fu, J.; Wang, H.; Wang, J.; Huang, C.; Prasanna, B.M.; Olsen, M.S.; Wang, G.; Zhang, A. Enhancing Genetic Gain through Genomic Selection: From Livestock to Plants. Plant Commun. 2020, 1, 100005. [Google Scholar] [CrossRef]
- Budhlakoti, N.; Kushwaha, A.K.; Rai, A.; Chaturvedi, K.K.; Kumar, A.; Pradhan, A.K.; Kumar, U.; Kumar, R.R.; Juliana, P.; Mishra, D.C.; et al. Genomic Selection: A Tool for Accelerating the Efficiency of Molecular Breeding for Development of Climate-Resilient Crops. Front. Genet. 2022, 13, 832153. [Google Scholar] [CrossRef]
- He, L.; Xiao, J.; Rashid, K.; Jia, G.; Li, P.; Yao, Z.; Wang, X.; Cloutier, S.; You, F. Evaluation of genomic prediction for pasmo resistance in flax. Int. J. Mol. Sci. 2019, 20, 359–376. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Hu, Z.L.; Xu, C.W. Genomic selection methods for crop improvement: Current status and prospects. Crop J. 2018, 6, 330–340. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Yan, J.; Zou, D.; Li, C.; Zhang, Z.; Song, S.; Wang, X. SR4R: An Integrative SNP Resource for Genomic Breeding and Population Research in Rice. Genom. Proteom. Bioinform. 2020, 18, 173–185. [Google Scholar] [CrossRef]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Peterson, D.; Tamura, K. MEGA-CC: Computing core of molecular evolutionary genetics analysis program for automated and iterative data analysis. Bioinformatics 2012, 28, 2685–2686. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.S.; He, W.M.; Ji, J.J.; Zhang, C.; Guo, Y.; Yang, T.L. LDBlockShow: A fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief. Bioinform. 2021, 22, bbaa227. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Y.W.; Xiang, Y.; Liu, M.H.; Zhang, Y.M. IIIVmrMLM: The R and C++ tools associated with 3VmrMLM, a comprehensive GWAS method for dissecting quantitative traits. Mol. Plant 2022, 15, 1251–1253. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, Z.; Wang, K.; Wang, R.; Fang, C. Comparative metabolomic analysis reveals the role of OsHPL1 in the cold-induced metabolic changes in rice. Plants 2023, 12, 2032. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Number | Range | Mean | SD | Skewness | Kurtosis | CV (%) a | H2 |
|---|---|---|---|---|---|---|---|---|
| Val-2012r1 | 422 | 20.34–24.67 | 22.28 | 0.63 | 0.55 | 0.77 | 53.65 | 0.45 |
| Val-2012r2 | 422 | 21.72–25.2 | 23.26 | 0.61 | 0.29 | 0.02 | 47.24 | |
| Val-2013r1 | 422 | 20.99–27.18 | 23.61 | 1.14 | 0.12 | −0.31 | 92.75 | |
| Val-2013r2 | 422 | 20.52–25.11 | 22.95 | 0.70 | 0.07 | 0.62 | 54.26 | |
| Leu-2012r1 | 422 | 7.02–14.59 | 10.78 | 2.09 | −0.81 | −0.52 | 99.42 | 0.49 |
| Leu-2012r2 | 422 | 9.85–15.82 | 12.98 | 0.91 | −0.11 | 0.09 | 67.34 | |
| Leu-2013r1 | 422 | 7.78–16.13 | 12.32 | 1.63 | −0.27 | −0.34 | 113.39 | |
| Leu-2013r2 | 422 | 9.67–14.72 | 12.24 | 0.92 | −0.10 | −0.05 | 66.93 | |
| Ile-2012r1 | 422 | 7.02–19.97 | 14.16 | 2.51 | −0.86 | 1.34 | 187.57 | 0.82 |
| Ile-2012r2 | 422 | 7.07–20.6 | 15.93 | 1.94 | −0.35 | 1.07 | 156.20 | |
| Ile-2013r1 | 422 | 9.06–22.33 | 15.95 | 2.13 | −0.09 | −0.04 | 219.03 | |
| Ile-2013r2 | 422 | 7.02–19.68 | 15.03 | 1.91 | −0.45 | 1.12 | 145.75 |
| QQI No. | Trait | QTN-1 | QTN-2 | aa.effect | LOD | p-Value | R2 (%) |
|---|---|---|---|---|---|---|---|
| QQI-1 | Leu | sf0206778257 | sf0427202052 | 0.25 | 4.80 | 2.61 × 10−6 | 4.50 |
| QQI-2 | Leu | sf0220165492 | sf0600825407 | 0.22 | 5.41 | 6.00 × 10−7 | 4.84 |
| QQI-3 | Leu | sf0414500497 | sf0810749006 | −0.19 | 5.47 | 5.14 × 10−7 | 3.90 |
| QQI-4 | Leu | sf0606055009 | sf1226791763 | −0.20 | 5.19 | 1.01 × 10−6 | 4.16 |
| QQI-5 | Leu | sf0608765681 | sf0815347583 | 0.30 | 5.02 | 1.52 × 10−6 | 5.84 |
| QQI-6 | Leu | sf0711167346 | sf0725292893 | 0.31 | 5.61 | 3.73 × 10−7 | 4.17 |
| QQI-7 | Leu | sf1109127718 | sf1207700947 | −0.29 | 4.99 | 1.63 × 10−6 | 3.67 |
| QQI-8 | Ile | sf0433008960 | sf1002475086 | −0.31 | 6.44 | 5.15 × 10−8 | 2.31 |
| QQI-9 | Ile | sf0622321011 | sf0701768847 | −0.29 | 5.79 | 2.42 × 10−7 | 2.21 |
| QQI-10 | Ile | sf0723859952 | sf1108192319 | 0.34 | 7.40 | 5.25 × 10−9 | 2.56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sui, Y.; Che, Y.; Zhong, Y.; He, L. Genome-Wide Association Studies Using 3VmrMLM Model Provide New Insights into Branched-Chain Amino Acid Contents in Rice Grains. Plants 2023, 12, 2970. https://doi.org/10.3390/plants12162970
Sui Y, Che Y, Zhong Y, He L. Genome-Wide Association Studies Using 3VmrMLM Model Provide New Insights into Branched-Chain Amino Acid Contents in Rice Grains. Plants. 2023; 12(16):2970. https://doi.org/10.3390/plants12162970
Chicago/Turabian StyleSui, Yao, Yanru Che, Yue Zhong, and Liqiang He. 2023. "Genome-Wide Association Studies Using 3VmrMLM Model Provide New Insights into Branched-Chain Amino Acid Contents in Rice Grains" Plants 12, no. 16: 2970. https://doi.org/10.3390/plants12162970
APA StyleSui, Y., Che, Y., Zhong, Y., & He, L. (2023). Genome-Wide Association Studies Using 3VmrMLM Model Provide New Insights into Branched-Chain Amino Acid Contents in Rice Grains. Plants, 12(16), 2970. https://doi.org/10.3390/plants12162970