1. Introduction
Natural disasters are becoming more often as the world’s climate changes, resulting in a significant drop in crop production. Among these various abiotic stresses, flooding stress has emerged as the greatest threat to crop growth and development, affecting crop production and yield worldwide [
1]. Due to excessive rainfall and poor drainage, the soil becomes flooded, causing hypoxia in below-ground parts and inhibiting plant growth [
2]. Soybeans (
Glycine max [L.] Merr.) are one of the most important commercial crops in the world due to its high nutritional values, containing protein, oil, carbohydrates, and isoflavones [
3]. However, soybeans are relatively susceptible to flooding stress, with considerable growth and yield inhibition under flood conditions [
4,
5]. Flooding stress affects soybeans at different growth and development stages, including germination, vegetative, and reproductive stages, which results in a great loss in soybean yields [
3]. Moreover, flooding imposes a severe selection pressure on plants since excess water in the living surroundings can deprive them of oxygen, carbon dioxide, and light [
6]. Flooding stress is considered as a primary limiting factor in soybean cultivation because 60–70% of annual precipitation occurs during the growth and development period [
7]. Additionally, several studies have displayed that flooding stress has a consistent impact on soybean growth and grain yields around the world, particularly in key soybean-producing countries such as China and United States [
8]. For example, in the Mississippi Delta region, flooding stress reduced overall soybean yield by up to 25% [
9]. Previous studies also focused on the understanding of flooding influences on different soybean growth stages and yield cutbacks. It is reported that flooding reduced soybean yields by 17–43% at the vegetative growth stage and 50–56% at the reproductive stage [
8,
10]. Similarly, Sullivan et al. (2001) exhibited a 20% yield loss when soybean plots were flooded for three days at V2 and V3 growth stages [
11].
Soybean flooding tolerance is considered to be a complex quantitative trait. Hence, QTL mapping is an effective way to unravel the underlying genetic mechanism of the tolerance. 27 QTLs on Soybase (
http://www.soybase.org) (accessed on 5 February 2023) have been reported to be linked to flooding tolerance at seedling stage in soybeans by linkage mapping approaches [
12,
13] For example, Zhang et al. [
14] detected nine significant QTLs for flooding tolerance on chromosomes 1, 4, 5, 16, and 18 for both flooding tolerance scores (FTSs) and survival rates (SRs) using a ‘Benning × PI 416937’ recombinant inbred line population. Over 20 QTLs were also found in another RIL population ‘Danbaekkong × NTS1116’ [
15,
16]. These QTLs are distributed across 20 chromosomes in the soybean genome, indicating the complex QTL architecture for the soybean tolerance. However, some major loci such as
qWT_Gm03 on chromosome 3 for waterlogging tolerance and
Qrld-12 for root length development (RLD) or
Qrsad-12 for root surface area development (RSAD) on chromosome 12 have been confirmed to promote the stress tolerance [
17,
18].
In contrast to waterlogging tolerance research of soybean plants with a focus on plant injury and yield loss, the effect of seed-flooding tolerance at germination stage has not been well determined. Due to the excessive rainfall at sowing time in some areas, soybean cultivars with seed-flooding tolerance at germination stage are desirable for breeding. A previous study found no significant differences in the evaluation of seed-flooding tolerance between laboratory and soil experiment [
19]. It is noteworthy that four integrated QTLs named
Sft1,
Sft2,
Sft3, and
Sft4 were identified by evaluating germination rate (GR) and normal seedling rate (NS) at germination stage.
Sft2 on Chromosome 08 exhibited large effects on seed-flooding tolerance. Interestingly, this QTL was near
I genetic locus which controls the seed coat pigmentation [
20]. A total of 33 QTLs including 26 main-effect ones for the seed-flooding tolerance parameter of relative seedling length were identified in two related RIL populations with a common tolerance parent M8206 by using a restricted two-stage multi-locus multi-allele genome-wide association study [
21]. By using a genome-wide association analysis (GWAS) method, 14 SNPs were found to be associated with the tolerance to waterlogging [
22]. A total of 25 QTNs associated with GR, NS, and electric conductivity (EC) were detected in a germplasm population [
23]. As one of the important indicators, the value of EC marked the intracellular substance leakage of soybean seeds under stress conditions, including flooding stress [
24]. EC was also found to be closely related to the seed vigor, which reflects the seed germination and unhealthy seedling developments [
25], and the germination test can be replaced by EC measurements carried out under soaking treatment [
26,
27]. Moreover, a total of 40 and 20 SNPs associated with GR and EC were found on a panel of 243 Plant Introduction from the USDA collection [
28]. However, these QTLs are detected from a limited cultivated soybean background. It is essential to mine and detect stable quantitative trait loci (QTLs) and related candidate genes in more genetic backgrounds.
Compared with cultivated soybean (
Glycine max (L.) Merr.), the annual wild soybean (
Glycine soja Sieb. and Zucc.) displayed high genetic diversity and is acknowledged as the progenitor of cultivated soybean [
29,
30]. Previous studies demonstrated that the wild soybean not only has abundant oil [
31] and protein content [
32] but also exhibits strong adaptability and resistance/tolerance to various biotic and abiotic stresses [
33,
34,
35,
36], indicating that wild soybean can be used as the important source of genetic variability. Thus, we can improve the seed-flooding tolerance of cultivated soybean by introducing favorable alleles from wild soybean into elite cultivars. Wild soybean accessions were shown to be more waterlog-tolerant than cultivated soybean in a recent study [
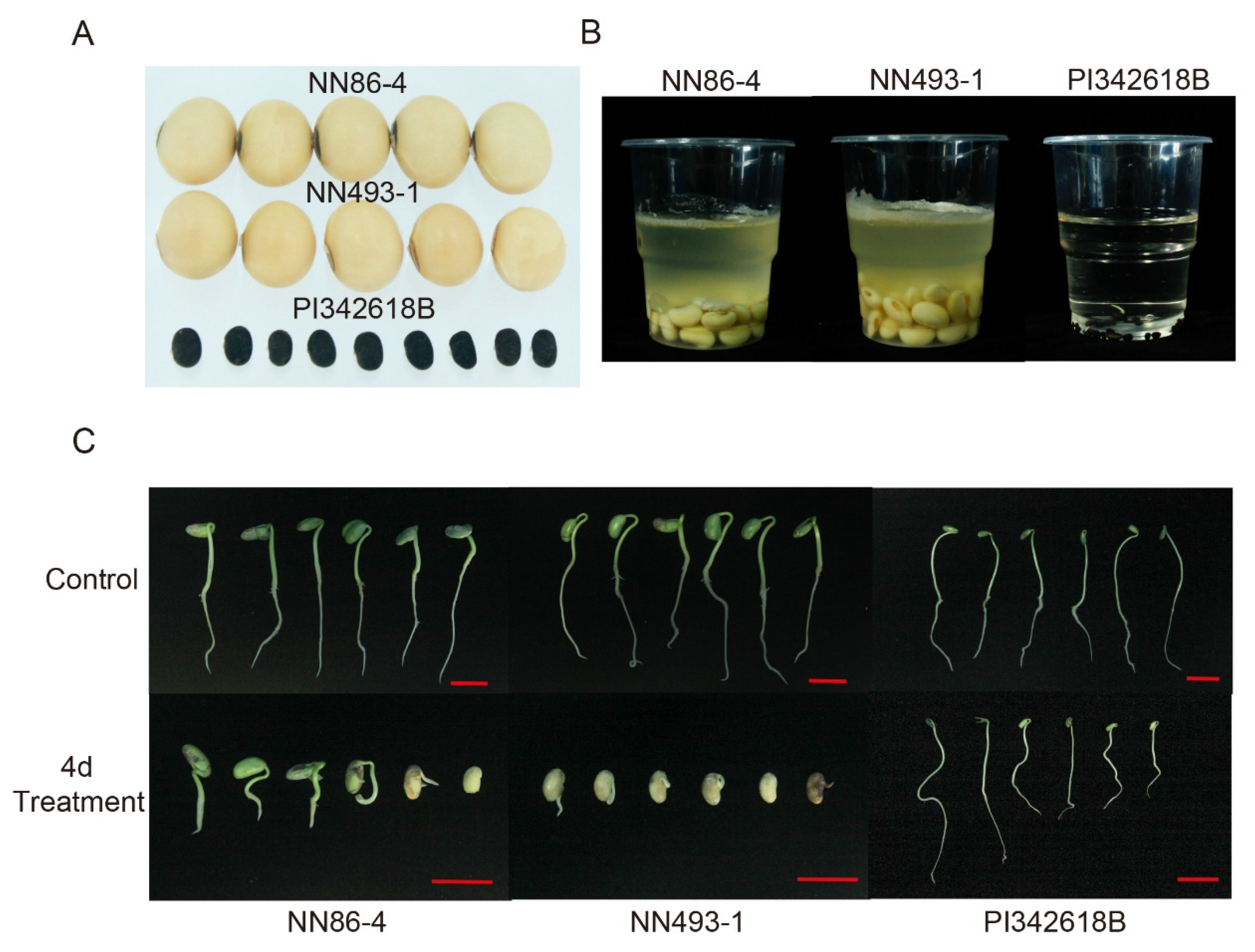
37]. Furthermore, the wild soybean accession PI342618B has demonstrated high seed-flooding tolerance [
38], with the majority of seedlings surviving after a 140 h flooding treatment, and five candidate genes for the tolerance were screened out from differentially expressed genes of root tissue transcriptomic profile between PI342618B and a sensitive line [
38]. By keeping the above in view, the objective of the present study was to identify QTLs for seed-flooding tolerance using two RIL populations derived by crossing common wild soybean accession PI342618B with two sensitive cultivated soybeans, NN86-4 and NN493-1, and then to mine candidate genes and elite alleles from
Glycine soja in developing high seed-flooding tolerant soybean varieties.
4. Discussion
Soybean was found to be relatively sensitive to flooding stress during germination, vegetative, and reproductive stages in a previous study [
52]. It is an effective way to reduce the impact of flooding stress on soybean production by screening elite flooding tolerant germplasm and breeding high tolerant varieties [
53]. However, the flooding tolerance of soybeans is a complex quantitative trait governed by multiple loci/genes with high environmental influence [
14,
20]. Over the past decades, 27 QTLs have been detected and reported on SoyBase using bi-parental mapping populations, which are distributed across the 20 chromosomes in the soybean genome, with several more recently detected [
14,
16]. Yu et al. (2019) identified 25 and 21 quantitative trait nucleotides (QTNs) by the mixed linear model (MLM) and multi-locus random-SNP-effect mixed linear model (mrMLM) of GWAS [
23], respectively. The two separate models for germination rate (GR), electrical conductivity (EC), and normal seedling rate (NSR) detected QTN13 on chromosome 13.
Glyma.13g248000 (
GmSFT), a QTN13-predicted candidate gene, was discovered to have a nonsynonymous mutation in seed-flooding tolerant genotypes, leading to an amino acid substitution in the protein. However, some major QTLs could be effectively utilized in breeding improved flooding-tolerant varieties or identifying candidate genes using marker-assisted selection (MAS). In this context, to better dissect the genetic basis of seed-flooding tolerance in soybean, two inter-specific high-density linkage maps of NJRINP and NJRI4P populations were utilized to identify major and stable QTLs related to seed-flooding tolerance using three parameters (GR, NSR, and EC) at the germination stage. Furthermore, two different methods, CIM and MCIM, were performed to improve the accuracy of mapping results. A total of 25 and 18 QTLs were detected by CIM and MCIM, respectively. Among these QTLs, 12 common QTLs were verified through both CIM and MCIM, indicating that these QTLs were stable and could be effectively utilized as potential candidate regions for enhancing seed-flooding tolerance. Interestingly, the mapping results displayed that QTLs related to various traits were distributed as clusters, and QTL hotspots of seed-flooding tolerance were identified on Chromosome 6 and Chromosome 8. We detected two QTL hotspots “6-1 and 6-2” on Chromosome 6. All the QTLs contained in hotspot “6-1”and hotspot “6-2” were only identified in a single population (NJIR4P) and explained small phenotypic variation with
R2 < 10.0%, indicating that these loci were minor QTLs related to seed-flooding tolerance.
Among the identified QTL hotspots, QTL hotspot “8-2” possessing QTLs associated with all three traits was detected in both populations and environments, while QTLs within hotspot “8-1”and hotspot “8-3” were only detectable in single populations (NJIR4P or NJIRNP). Furthermore, QTL hotspot “8-2” was validated by both CIM and MCIM, and all the QTLs contained in “hotspot 8-2” displayed large effects on seed-flooding tolerance (
R2 > 10.0%). Some previous studies also displayed major QTLs related to seed-flooding tolerance in this map region [
20,
28]. For example, Sayama et al. (2009) identified
sft2 near the
I locus on chromosome 8 (LG_A2) [
20], which is involved in seed coat pigmentation. Therefore, it was concluded that QTL hotspot “8-2” was the most likely candidate region for seed-flooding tolerance. Besides the QTL hotspots detected on Chromosome 6 and Chromosome 8, we also identified some QTLs on other chromosomes, including Chromosome 1, Chromosome 3, Chromosome 15, and so on. By comparing the genomic position of these QTLs with the previously identified loci,
qGR-1-1 and
qEC-1-1 are overlapped with the
Flood tolerance 6-1 locus coded on the SoyBase website detected by Rizal and Karki [
54], and the region of
qEC-17-1 on Chromosome 17 also includes
QTL SFT_17-52 and
RSL-a-17-1a found by Dhungana et al. [
16] and Ali et al. [
21].
The genetic makeup of complex traits is not only regulated by main-effect QTLs/genes but also by inter-locus interactions as well as their interactions with the environment [
55]. Understanding the epistasis and QTL × environment (QE) interaction effects are essential for revealing the crucial genetic basis for determining quantitative traits and improving QTL detection and selection accuracy. Hence, ignoring inter-genic interaction will result in an overestimation of individual QTL effects and an underestimation of genetic variance [
56]. In this study, four pairs of digenic epistatic QTLs were detected, three for EC and one for NSR. Out of these, only one epistatic QTL pair (
qEC-4-1 and
qEC-8-1) displayed individual additive effects, while the remaining three epistatic QTL pairs showed non-additive effects. These three epistatic QTL pairs might serve as modifying genes that themselves have no significant effects but regulate the expression of flooding-tolerance related genes through epistatic interactions. All four pairs have significant AA effects. However, the total PVE explained by four epistatic pairs related to seed-flooding tolerance was about 14.19%. Similar results for the epistatic interaction of QTLs for different traits in soybean have been reported in early studies [
57,
58]. The presence of epistatic interactions for a given trait makes selection more difficult. Interestingly, all main-effect QTLs identified in the present study, except for
qEC-4-1 and
qEC-8-1, had no epistatic interactions, which increased the heritability of the trait and made selection easier.
A previous study found a strong correlation between seed-flooding tolerance and seed-coat color in soybean [
19]. Moreover, early study also demonstrated that the seed-flooding of pigmented genotypes was significantly more tolerant compared to genotypes with yellow seed-coat [
20]. Therefore, these results potentially indicated that
Sft2 and
I were the same locus or that they possessed strong linkage with each other [
20]. Similar results were observed in the present study that genotypes with pigmented seed-coat (black, brown, green, and other pigmented colors) exhibited more seed-flooding tolerance than those with yellow seed-coat in NJIRNP and NJIR4P populations. These findings provide further evidence that the seed-coat pigmentation is strongly associated with seed-flooding tolerance. Interestingly, three identified QTL “hotspots 8-1, 8-2 and 8-3” for seed-flooding tolerance in our study were located near the
I locus on Chromosome 8. It is known that the
I locus controlling seed-coat pigmentation encompasses a region of three duplicated chalcone synthase genes (
CHS1,
CHS3, and
CHS4) in soybean [
59]. Thus, to exclude the possibility that these three
CHS genes were the candidate genes involved in seed-flooding tolerance, we performed the sequence analysis of
CHS genes between PI342616B and NN86-4, NN 493-1. The results revealed no changes were observed in the amino acid sequence of
CHS proteins among parents, indicating that the
CHS pathway may be independent of seed-flooding tolerance. Furthermore, our additional evidence supported this viewpoint because we observed many accessions with the yellow seed-coat exhibiting high GR and NSR in both populations in the present study.
So far, limited studies have been carried out to identify candidate genes for seed-flooding tolerance in soybean. In this regard, mining of the candidate genes for seed-flooding tolerance in soybean revealed 164 model genes within the physical intervals of QTL “hotspot 8-2” on Chromosome 8 in the present study. Based on the expression levels of these genes in different soybean tissues from Soybase (
https://www.soybase.org/soyseq/) and the functional annotations related to seed-flooding tolerance, ten candidate genes were screened. The qRT-PCR and sequence analysis were further performed, and the results revealed that
Glyma.08G137600 was significantly induced under 4 d of flooding stress in all three parents (PI342618B, NN86-4, and NN493-1) compared with the control. Moreover, three consecutive base insertions (-/TTC) in the coding region of PI342616B were observed, and this mutation resulted in the insertion of serine in the low complexity region of protein. These results indicated that
Glyma.08G137600 was the most likely possible candidate gene for seed-flooding tolerance in soybean, and this gene was designated as
GmDREB2. The functional annotation of
GmDREB2 exhibited that it encodes the ERF transcription factor, which contains the AP2 domain. In rice, an ethylene response factor (ERF) gene named
Sub1A was characterized and involved in submergence tolerance [
60]. Moreover, two ethylene response factors
SNORKEL1 and
SNORKEL2, which play vital roles in response to deepwater stress by ethylene signaling, were identified [
61]. In Arabidopsis, it was demonstrated that three ERF-ⅦII transcription factors (
RAP2.2,
RAP2.3, and
RAP2.12) were involved in regulating and activating the core anaerobic response [
62]. All the above functional studies illustrate the important role of the ERF transcription factor in flooding stress. Thus,
GmDREB2 was considered the most likely candidate gene for seed-flooding tolerance in soybean in the present study. The results demonstrated that the overexpression of
GmDREB2 relatively promoted the growth of soybean hairy roots under flooding stress, indicating that
GmDREB2 was positively involved in the regulation of seed-flooding tolerance in soybean. However, it requires more verification experiments to validate the molecular function of
GmDREB2 for its specific roles in seed-flooding tolerance.
In the present study, we reported that the wild parent PI342616B exhibited strong seed-flooding tolerance compared to the sensitive cultivar parents NN86-4 and NN493-1 under flooding stress. Moreover, all the favorable positive alleles of identified QTLs were derived from PI342616B, which supported the view that wild soybean gene pool is an abundant source of elite stress-tolerant genes. Wild soybean, as the progenitor of cultivated soybean, has been reported to display high contents of oil and protein, rich in genetic diversity, strong adaptability, and tolerance to various stresses [
21,
22,
23]. Due to the favorable tolerance alleles contributed by wild soybean, it has become an efficient way to improve the seed-flooding tolerance of elite soybean varieties by introducing the seed-flooding tolerant QTLs/genes from wild soybean into elite cultivars.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}