

A Fast and Cost-Effective Genotyping Method for CRISPR-Cas9-Generated Mutant Rice Lines

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Direct PCR Amplification from Plant Tissues Is as Efficient as from Extracted DNA
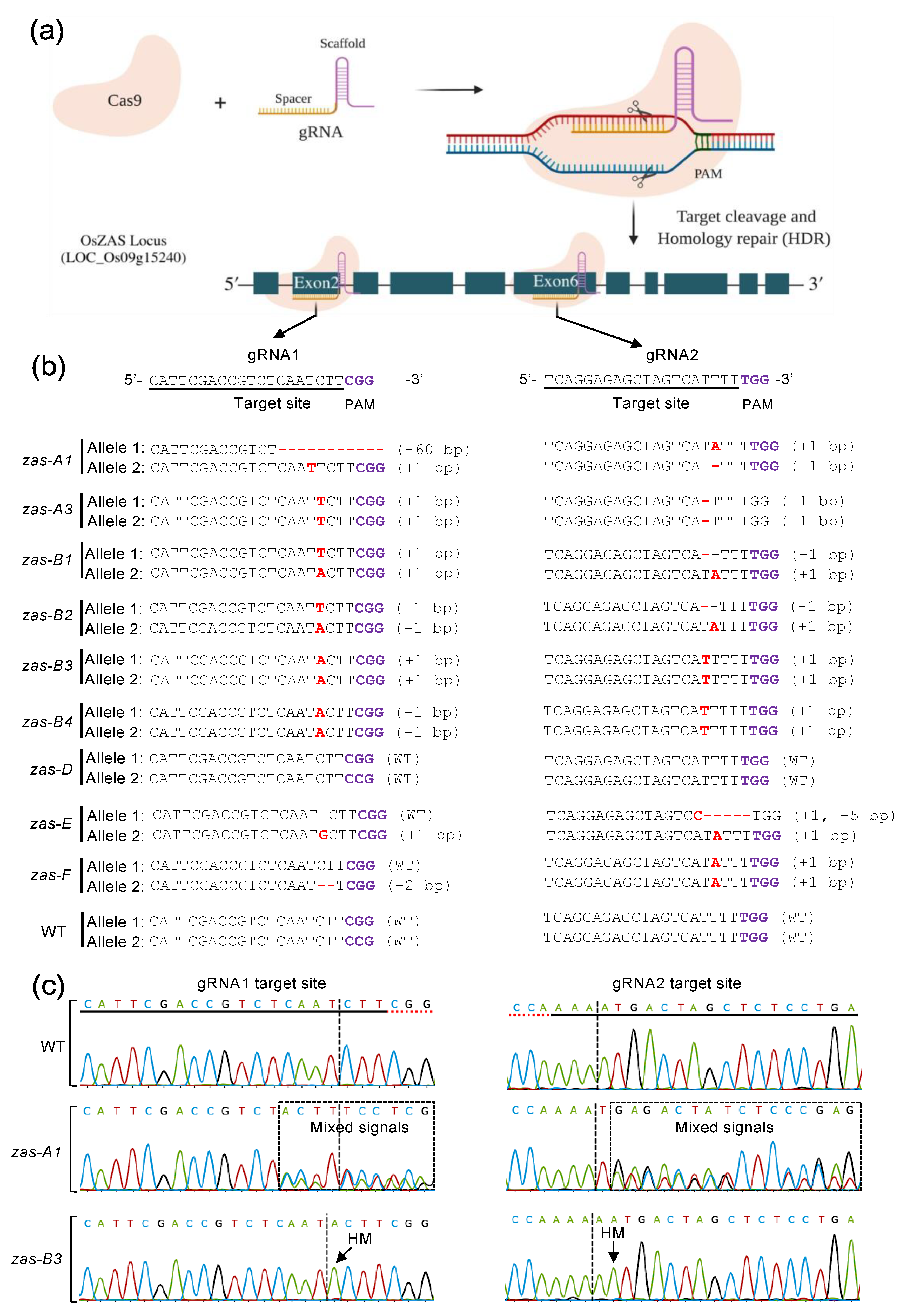
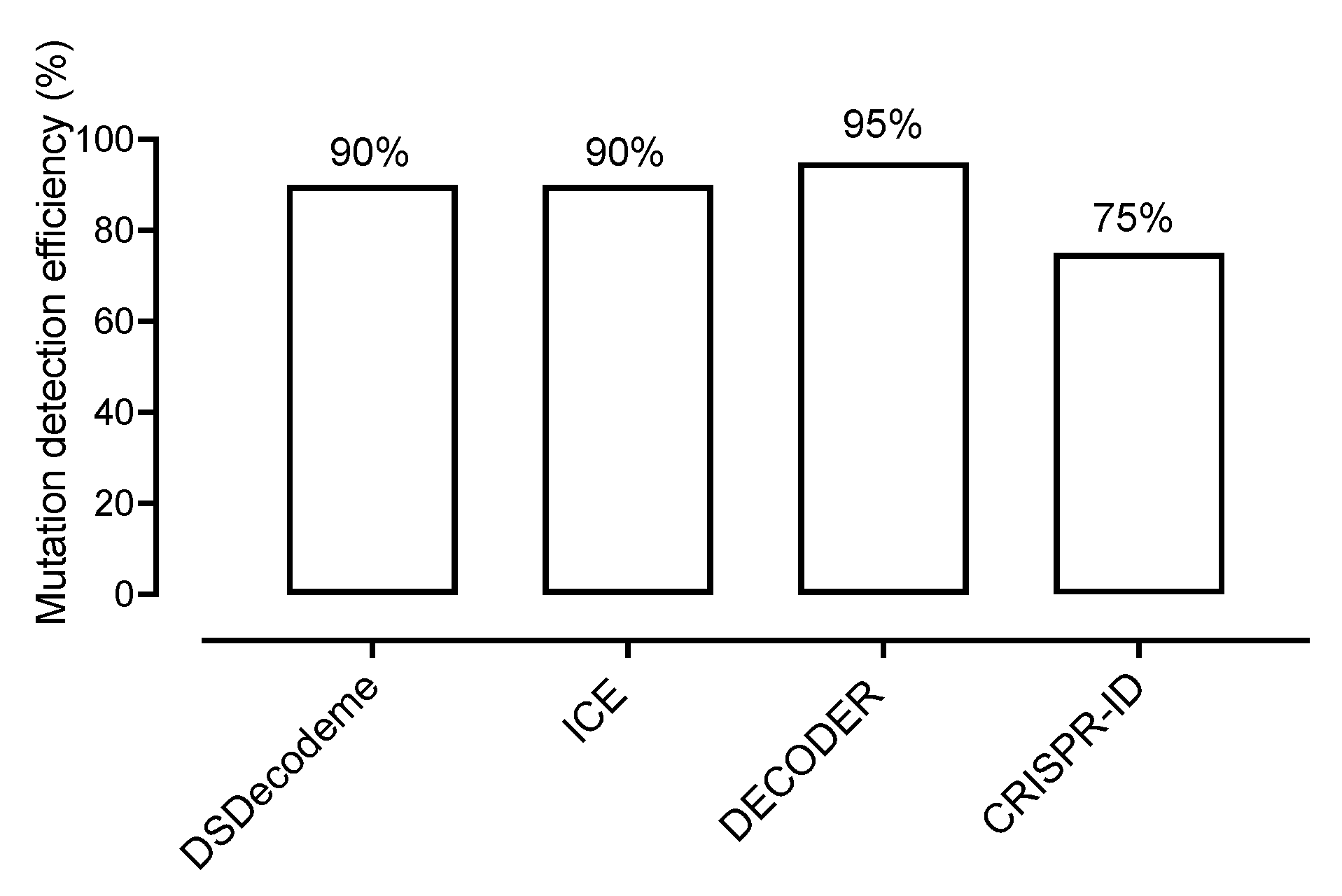
2.2. Free Online, Automatic Analysis of CRISPR-Induced Mutations Is Mostly Accurate
2.3. Proposed Workflow Much Faster and Cheaper
3. Material and Methods
3.1. CRISPR Targets and Vector Construction
3.2. Rice Transformation
3.3. Transgenicity and Mutagenicity Analysis
3.4. Traditional Workflow
3.5. Proposed Workflow
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chauhan, B.S.; Jabran, K.; Mahajan, G. (Eds.) Rice Production Worldwide; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Izawa, T.; Shimamoto, K. Becoming a model plant: The importance of rice to plant science. Trends Plant Sci. 1996, 1, 95–99. [Google Scholar] [CrossRef]
- Hirochika, H.; Guiderdoni, E.; An, G.; Hsing, Y.-I.; Eun, M.Y.; Han, C.-D.; Upadhyaya, N.M.; Ramachandran, S.; Zhang, Q.; Pereira, A.; et al. Rice Mutant Resources for Gene Discovery. Plant Mol. Biol. 2004, 54, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Shan, Q.; Wang, Y.; Li, J.; Zhang, Y.; Chen, K.; Liang, Z.; Zhang, K.; Liu, J.; Xi, J.J.; Qiu, J.-L.; et al. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 686–688. [Google Scholar] [CrossRef]
- Miao, J.; Guo, D.; Zhang, J.; Huang, Q.; Qin, G.; Zhang, X.; Wan, J.; Gu, H.; Qu, L.-J. Targeted mutagenesis in rice using CRISPR-Cas system. Cell Res. 2013, 23, 1233–1236. [Google Scholar] [CrossRef]
- Hiei, Y.; Komari, T. Agrobacterium-mediated transformation of rice using immature embryos or calli induced from mature seed. Nat. Protoc. 2008, 3, 824–834. [Google Scholar] [CrossRef]
- Yue, J.-J.; Hong, C.-Y.; Wei, P.; Tsai, Y.-C.; Lin, C.-S. How to start your monocot CRISPR/Cas project: Plasmid design, efficiency detection, and offspring analysis. Rice 2020, 13, 9. [Google Scholar] [CrossRef]
- Romero, F.; Gatica-Arias, A. CRISPR/Cas9: Development and Application in Rice Breeding. Rice Sci. 2019, 26, 265–281. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Haeussler, M.; Concordet, J.-P. Genome Editing with CRISPR-Cas9: Can It Get Any Better? J. Genet. Genom. 2016, 43, 239–250. [Google Scholar] [CrossRef]
- Li, H.; Li, J.; Cong, X.; Duan, Y.; Li, L.; Wei, P.; Lu, X.; Yang, J. A high-throughput, high-quality plant genomic DNA extraction protocol. Genet. Mol. Res. 2013, 12, 4526–4539. [Google Scholar] [CrossRef]
- Yang, N.; Wang, R.; Zhao, Y. Revolutionize Genetic Studies and Crop Improvement with High-Throughput and Genome-Scale CRISPR/Cas9 Gene Editing Technology. Mol. Plant 2017, 10, 1141–1143. [Google Scholar] [CrossRef]
- Anderson, C.B.; Franzmayr, B.K.; Hong, S.W.; Larking, A.C.; van Stijn, T.C.; Tan, R.; Moraga, R.; Faville, M.J.; Griffiths, A.G. Protocol: A versatile, inexpensive, high-throughput plant genomic DNA extraction method suitable for genotyping-by-sequencing. Plant Methods 2018, 14, 75. [Google Scholar] [CrossRef]
- Biswas, S.; Li, R.; Yuan, Z.; Zhang, D.; Zhao, X.; Shi, J. Development of methods for effective identification of CRISPR/Cas9-induced indels in rice. Plant Cell Rep. 2019, 38, 503–510. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, C.; Jiao, X.; Zhang, H.; Song, L.; Li, Y.; Gao, C.; Wang, K. Hi-TOM: A platform for high-throughput tracking of mutations induced by CRISPR/Cas systems. Sci. China Life Sci. 2019, 62, 1–7. [Google Scholar] [CrossRef]
- Wang, J.Y.; Haider, I.; Jamil, M.; Fiorilli, V.; Saito, Y.; Mi, J.; Baz, L.; Kountche, B.A.; Jia, K.-P.; Guo, X.; et al. The apocarotenoid metabolite zaxinone regulates growth and strigolactone biosynthesis in rice. Nat. Commun. 2019, 10, 810. [Google Scholar] [CrossRef]
- Ablazov, A.; Mi, J.; Jamil, M.; Jia, K.P.; Wang, J.Y.; Feng, Q.; Al-Babili, S. The apocarotenoid zaxinone is a positive regulator of strigolactone and abscisic acid biosynthesis in Arabidopsis roots. Front. Plant Sci. 2020, 11, 578. [Google Scholar] [CrossRef]
- Wang, J.Y.; Alseekh, S.; Xiao, T.; Ablazov, A.; de Souza, L.P.; Fiorilli, V.; Anggarani, M.; Lin, P.-Y.; Votta, C.; Novero, M.; et al. Multi-omics approaches explain the growth-promoting effect of the apocarotenoid growth regulator zaxinone in rice. Commun. Biol. 2021, 4, 1222. [Google Scholar] [CrossRef]
- Votta, C.; Fiorilli, V.; Haider, I.; Wang, J.Y.; Balestrini, R.; Petřík, I.; Tarkowska, D.; Novak, O.; Serikbayeva, A.; Bonfante, P.; et al. Zaxinone synthase controls arbuscular mycorrhizal colonization level in rice. Plant J. 2022, 111, 1688–1700. [Google Scholar] [CrossRef]
- Ablazov, A.; Votta, C.; Fiorilli, V.; Wang, J.Y.; Aljedaani, F.; Jamil, M.; Balakrishna, A.; Balestrini, R.; Liew, K.X.; Rajan, C.; et al. ZAXINONE SYNTHASE 2 regulates growth and arbuscular mycorrhizal symbiosis in rice. Plant Physiol. 2022, 191, 382–399. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Kim, C.S.; Lee, C.H.; Shin, J.S.; Chung, Y.S.; Hyung, N.I. A Simple and Rapid Method for Isolation of High-Quality Genomic DNA from Fruit Trees and Conifers Using PVP. Nucleic Acids Res. 1997, 25, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Bellstedt, D.U.; Pirie, M.D.; Visser, J.C.; de Villiers, M.J.; Gehrke, B. A rapid and inexpensive method for the direct PCR amplification of DNA from plants. Am. J. Bot. 2010, 97, e65–e68. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.G.; Kim, J.Y.; Soh, M.-S.; Kim, O.-S. A Simple and Rapid Gene Amplification from Arabidopsis Leaves Using AnyDirect System. BMB Rep. 2007, 40, 444–447. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, H.; Yan, X.; Li, M.; Chen, P.; Zhang, S. A universal method for direct PCR amplification of plant tissues. Anal. Methods 2017, 9, 1800–1805. [Google Scholar] [CrossRef]
- Bell, J.R. A simple way to treat PCR products prior to sequencing using ExoSAP-IT®. Biotechniques 2008, 44, 834. [Google Scholar] [CrossRef]
- Lynagh, P.G.; Osuna-Kleist, P.; Wang, B.; Malagon, E.; Anleu Gil, M.X.; Comai, L. Accurate Direct PCR with Arabidopsis and rice. Plant Cell Physiol. 2023, 64, 1–3. [Google Scholar] [CrossRef]
- Liu, W.; Xie, X.; Ma, X.; Li, J.; Chen, J.; Liu, Y.-G. DSDecode: A Web-Based Tool for Decoding of Sequencing Chromatograms for Genotyping of Targeted Mutations. Mol. Plant 2015, 8, 1431–1433. [Google Scholar] [CrossRef]
- Conant, D.; Hsiau, T.; Rossi, N.; Oki, J.; Maures, T.; Waite, K.; Yang, J.; Joshi, S.; Kelso, R.; Holden, K.; et al. Inference of CRISPR Edits from Sanger Trace Data. CRISPR J. 2022, 5, 123–130. [Google Scholar] [CrossRef]
- Bloh, K.; Kanchana, R.; Bialk, P.; Banas, K.; Zhang, Z.; Yoo, B.-C.; Kmiec, E.B. Deconvolution of Complex DNA Repair (DECODR): Establishing a Novel Deconvolution Algorithm for Comprehensive Analysis of CRISPR-Edited Sanger Sequencing Data. CRISPR J. 2021, 4, 120–131. [Google Scholar] [CrossRef]
- Dehairs, J.; Talebi, A.; Cherifi, Y.; Swinnen, J.V. CRISP-ID: Decoding CRISPR mediated indels by Sanger sequencing. Sci. Rep. 2016, 6, 28973. [Google Scholar] [CrossRef]
- Xie, K.; Minkenberg, B.; Yang, Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc. Natl. Acad. Sci. USA 2015, 112, 3570–3575. [Google Scholar] [CrossRef]
- Dellaporta, S.L.; Wood, J.; Hicks, J.B. A Plant DNA Minipreparation: Version II. Plant Mol. Biol. Rep. 1983, 1, 19–21. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Lines | pJet Cloning | DSDecodeme | ICE Software | DECODER v3.0 | CRISPR-ID |
|---|---|---|---|---|---|---|
| 1 | zas-A1_gRNA1 | Biallelic mutation (HE) | correct | Single allele correct | correct | correct |
| 2 | zas-A3_gRNA1 | Homozygote (HO) | correct | correct | correct | correct |
| 3 | zas-B1_gRNA1 | Biallelic mutation (HE) | correct | correct | correct | not correct |
| 4 | zas-B2_gNA1 | Biallelic mutation (HE) | correct | correct | correct | not correct |
| 5 | zas-B3_gRNA1 | Homozygote (HO) | correct | correct | correct | correct |
| 6 | zas-B4_gRNA1 | Homozygote (HO) | correct | correct | correct | correct |
| 7 | zas-D_gRNA1 | No mutation | correct | correct | correct | not correct |
| 8 | zas-E_gRNA1 | Monoalleleci (HE) | correct | correct | correct | not correct |
| 9 | zas-F_gRNA1 | Monoallelic (HE) | Single allele correct | Single allele correct | Single allele correct | not correct |
| 10 | WT_gRNA1 | No mutation | correct | correct | correct | correct |
| 11 | zas-A1_gRNA2 | Biallelic mutation (HE) | correct | correct | correct | correct |
| 12 | zas-A3_gRNA2 | Homozygote (HO) | correct | correct | correct | correct |
| 13 | zas-B1_gRNA2 | Biallelic mutation (HE) | correct | correct | correct | correct |
| 14 | zas-B2_gNA2 | Biallelic mutation (HE) | correct | correct | correct | correct |
| 15 | zas-B3_gRNA2 | Homozygote (HO) | correct | correct | correct | correct |
| 16 | zas-B4_gRNA2 | Homozygote (HO) | correct | correct | correct | correct |
| 17 | zas-D_gRNA2 | No mutation | correct | correct | correct | correct |
| 18 | zas-E_gRNA2 | Biallelic mutation (HE) | decoding failed | correct | correct | correct |
| 19 | zas-F_gRNA2 | Homozygote (HO) | correct | correct | correct | correct |
| 20 | WT_gRNA2 | No mutation | correct | correct | correct | correct |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ablazov, A.; Felemban, A.; Braguy, J.; Kuijer, H.N.J.; Al-Babili, S. A Fast and Cost-Effective Genotyping Method for CRISPR-Cas9-Generated Mutant Rice Lines. Plants 2023, 12, 2189. https://doi.org/10.3390/plants12112189
Ablazov A, Felemban A, Braguy J, Kuijer HNJ, Al-Babili S. A Fast and Cost-Effective Genotyping Method for CRISPR-Cas9-Generated Mutant Rice Lines. Plants. 2023; 12(11):2189. https://doi.org/10.3390/plants12112189
Chicago/Turabian StyleAblazov, Abdugaffor, Abrar Felemban, Justine Braguy, Hendrik N. J. Kuijer, and Salim Al-Babili. 2023. "A Fast and Cost-Effective Genotyping Method for CRISPR-Cas9-Generated Mutant Rice Lines" Plants 12, no. 11: 2189. https://doi.org/10.3390/plants12112189
APA StyleAblazov, A., Felemban, A., Braguy, J., Kuijer, H. N. J., & Al-Babili, S. (2023). A Fast and Cost-Effective Genotyping Method for CRISPR-Cas9-Generated Mutant Rice Lines. Plants, 12(11), 2189. https://doi.org/10.3390/plants12112189