
Analysis of Comparative Transcriptome and Positively Selected Genes Reveal Adaptive Evolution in Leaf-Less and Root-Less Whisk Ferns

 ,
,
Abstract
:1. Introduction
2. Results
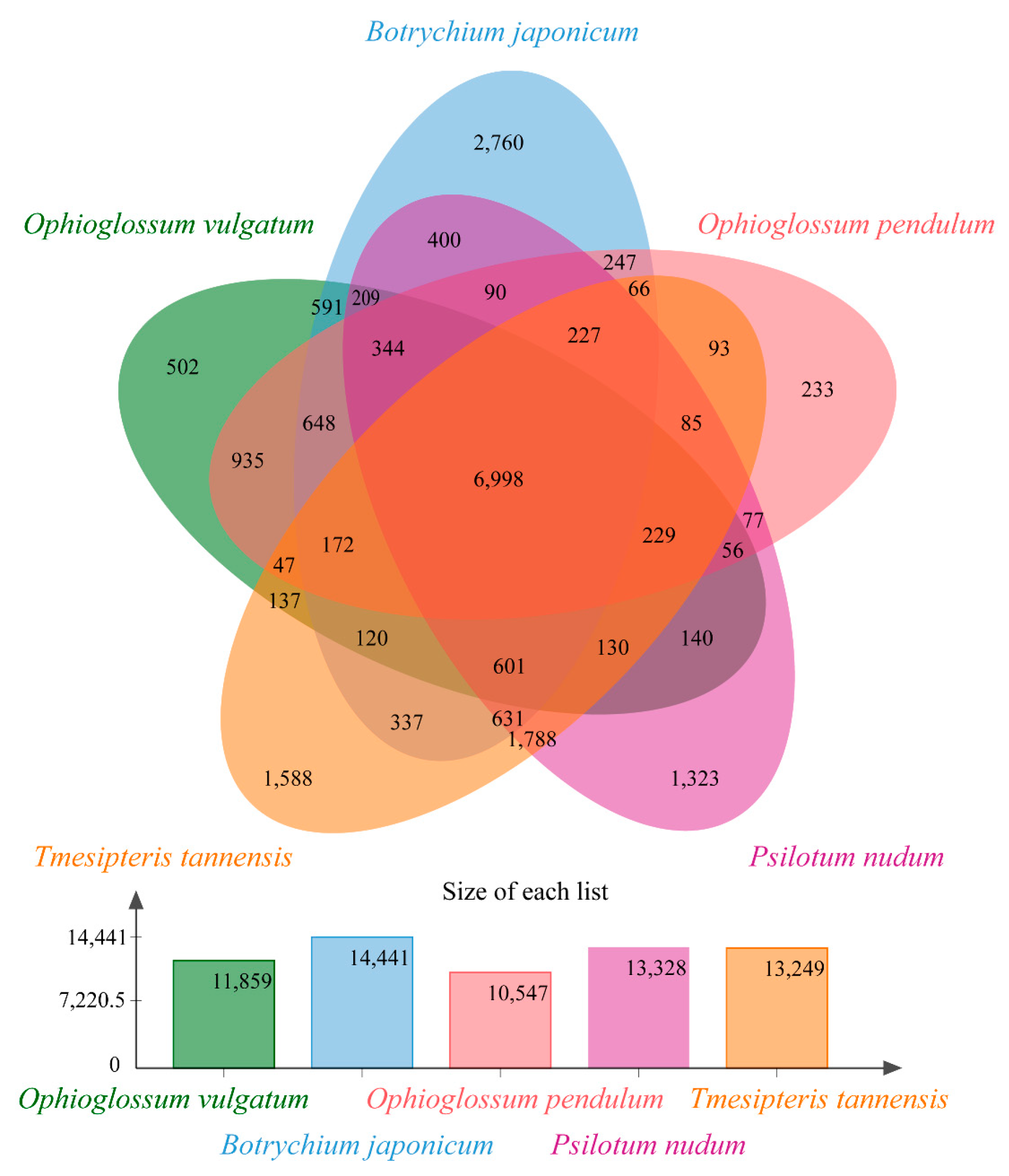
2.1. Assessment of Transcriptomes and Homolog Clusters
2.2. Comparative Transcriptome Analyses and Functional Enrichment
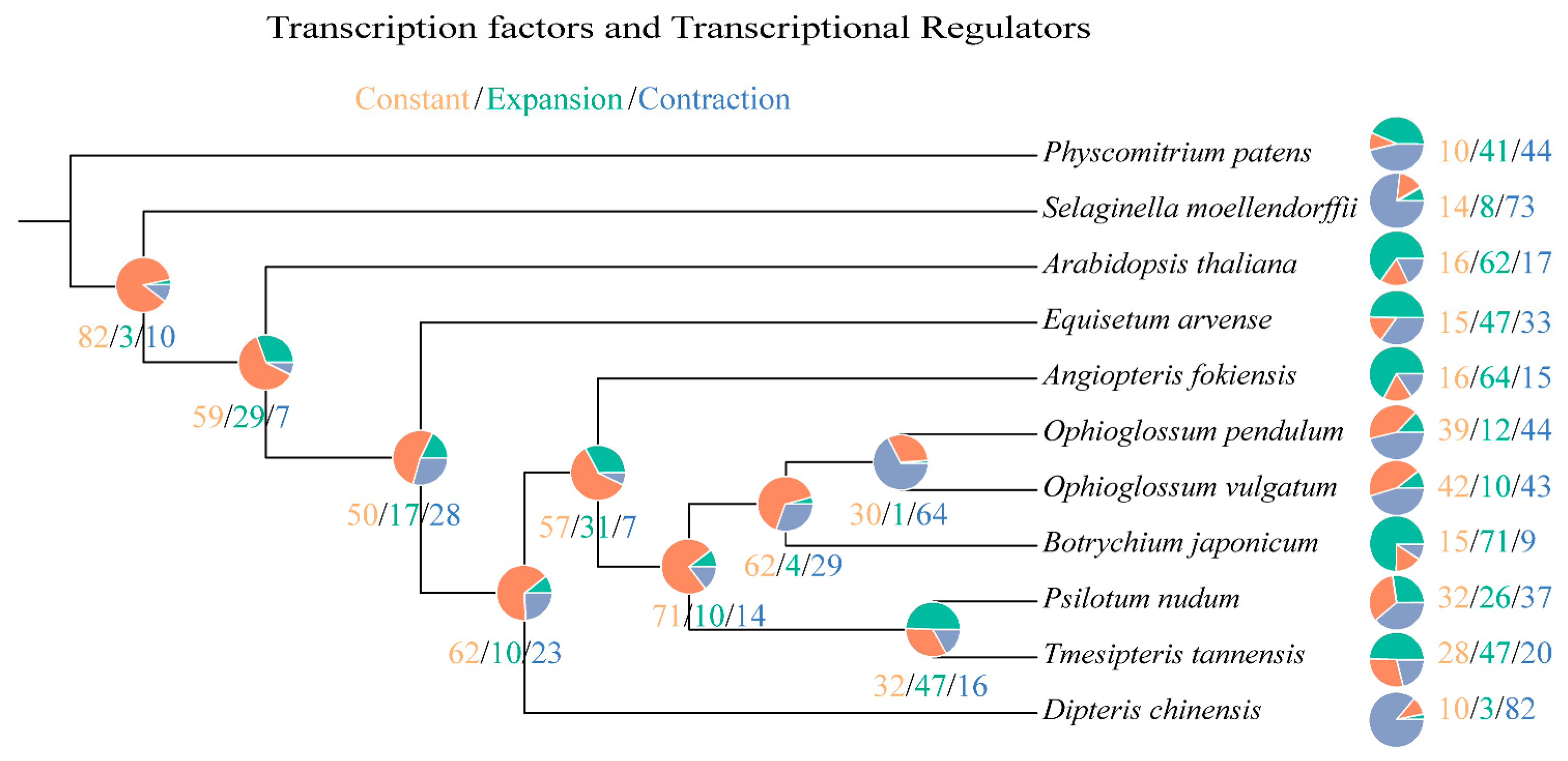
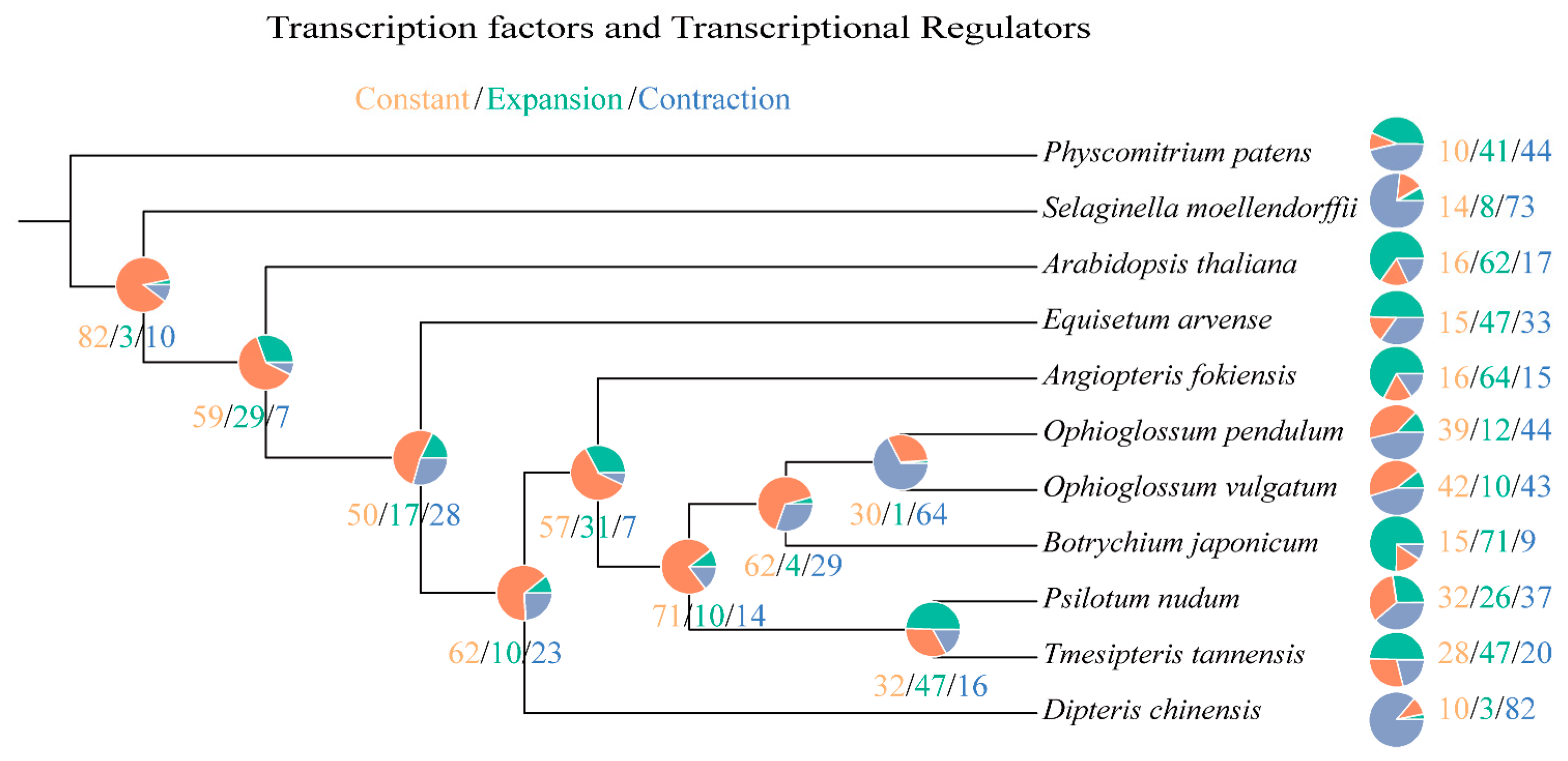
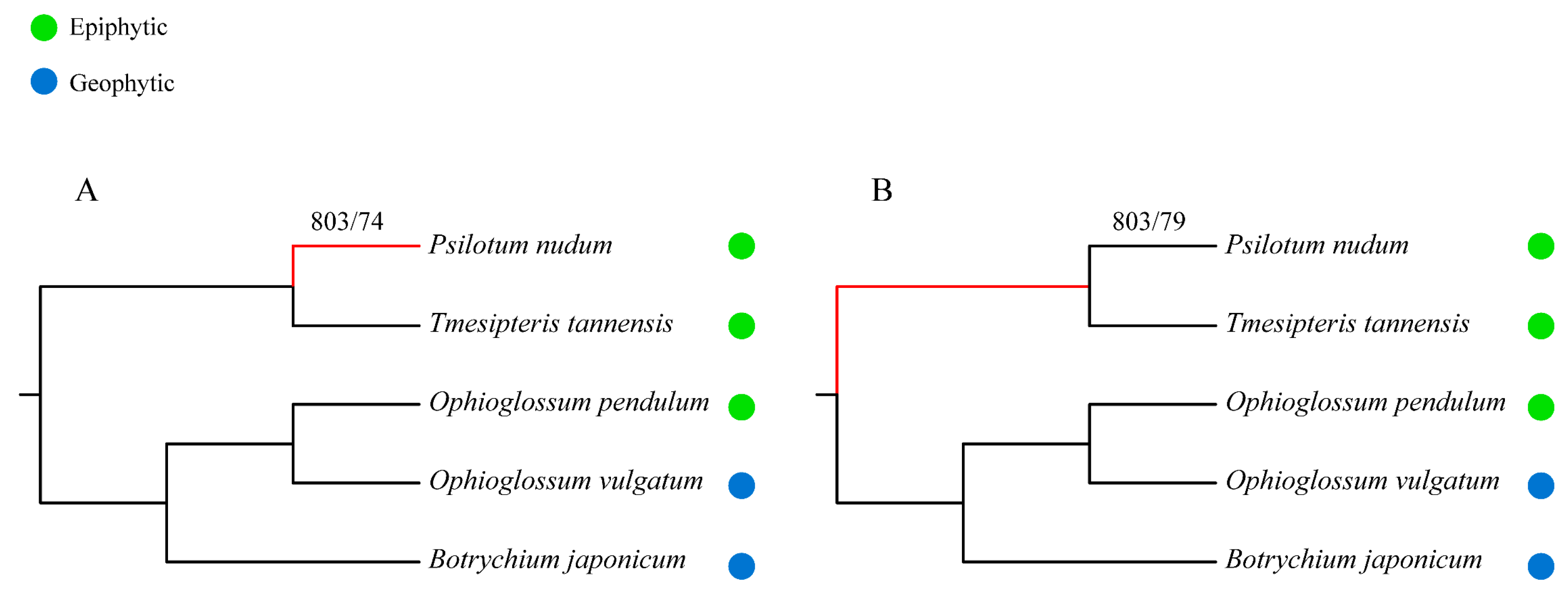
2.3. Phylogeny Reconstruction and Analysis on TF/TR Variation Related to Morphological Development
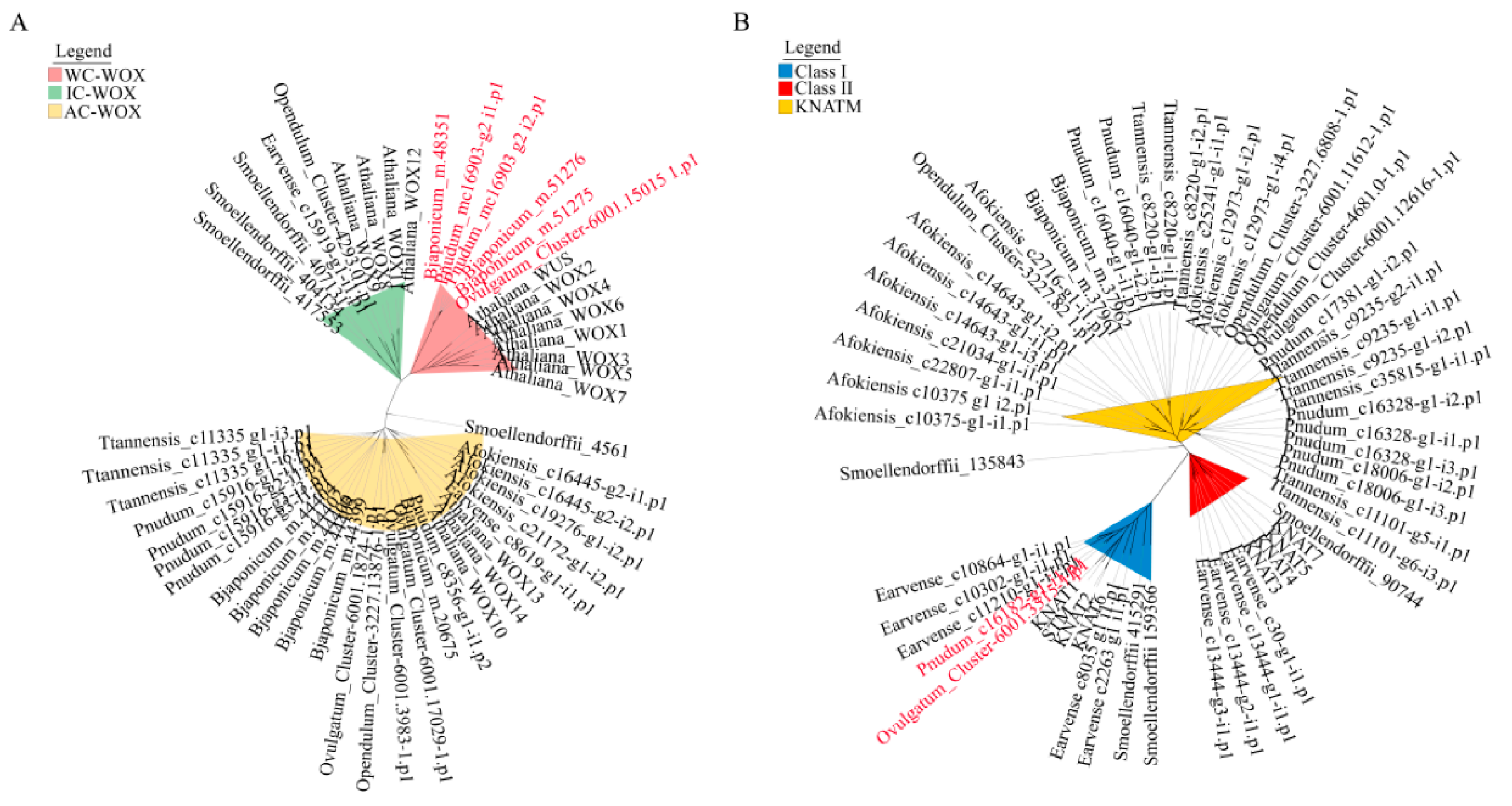
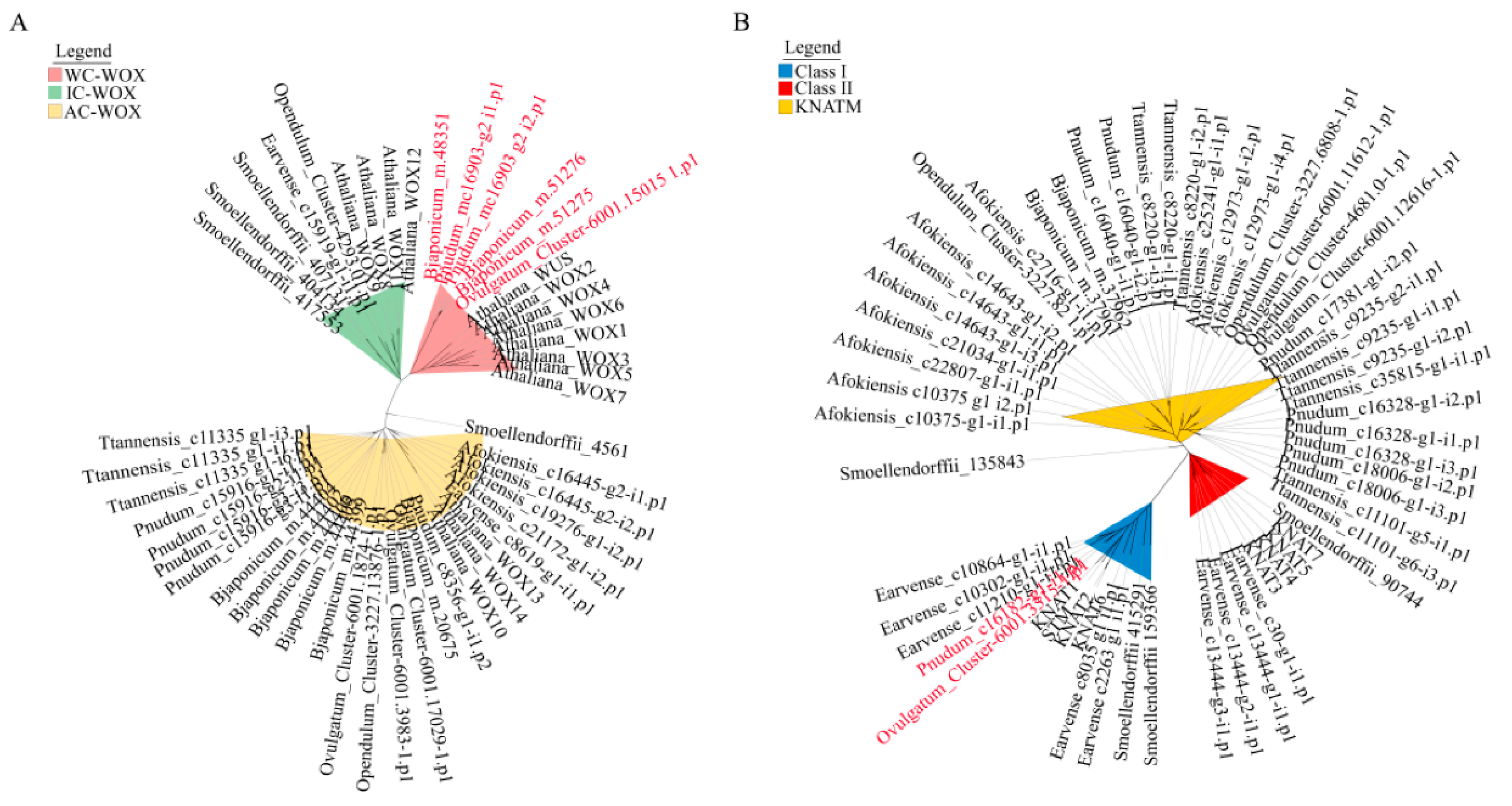
2.4. Phylogenetic Analysis of WOX and KNOX Genes
2.5. Genes under Positive Selection Detected in Whisk Ferns
3. Discussion
4. Materials and Methods
4.1. Transcriptome Data Collection and BUSCO Analysis
4.2. Phylogenetic Analysis
4.3. Comparative Transcriptome Analysis
4.4. Identification and Gene Tree Reconstruction of the WOX and KNOX Families
4.5. Detection of Genes under Positive Selection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haberlandt, G. Physiological Plant Anatomy; Macmillan: London, UK, 1914. [Google Scholar]
- Boyce, C.K.; Knoll, A.H. Evolution of developmental potential and the multiple independent origins of leaves in Paleozoic vascular plants. Paleobiology 2002, 28, 70–100. [Google Scholar] [CrossRef]
- Floyd, S.K.; Bowman, J.L. Distinct developmental mechanisms reflect the independent origins of leaves in vascular plants. Curr. Biol. 2006, 16, 1911–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenrick, P.; Strullu-Derrien, C. The origin and early evolution of roots. Plant Physiol. 2014, 166, 570–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Xu, L. Recruitment of IC-WOX genes in root evolution. Trends Plant Sci. 2018, 23, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Pires, N.D.; Dolan, L. Morphological evolution in land plants: New designs with old genes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 508–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raven, J.A.; Edwards, D. Roots: Evolutionary origins and biogeochemical significance. J. Exp. Bot. 2001, 52, 381–401. [Google Scholar] [CrossRef]
- Sanders, H.; Rothwell, G.W.; Wyatt, S.E. Key morphological alterations in the evolution of leaves. Int. J. Plant Sci. 2009, 170, 860–868. [Google Scholar] [CrossRef]
- Tomescu, A.M.F. Megaphylls, microphylls and the evolution of leaf development. Trends Plant Sci. 2009, 14, 5–12. [Google Scholar] [CrossRef]
- Ge, Y.; Fang, X.; Liu, W.; Sheng, L.; Xu, L. Adventitious lateral rooting: The plasticity of root system architecture. Physiol. Plant 2019, 165, 39–43. [Google Scholar] [CrossRef]
- Foster, A.S.; Gifford, E.M. Comparative Morphology of Vascular Plants; W. H. Freeman and Company: San Francisco, CA, USA, 1974. [Google Scholar]
- Schneider, H. Evolutionary morphology of ferns (monilophytes). Annu. Plant Rev. 2013, 45, 115–140. [Google Scholar]
- Taylor, E.L.; Taylor, T.N.; Krings, M. Paleobotany: The Biology and Evolution of Fossil Plants; Academic Press: Cambridge, MA, USA, 2009. [Google Scholar]
- PPG I.(The Pteridophyte Phylogeny Group). A community-derived classification for extant lycophytes and ferns. J. Syst. Evol. 2016, 54, 563–603. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hake, S.; Smith, H.M.; Holtan, H.; Magnani, E.; Mele, G.; Ramirez, J. The role of knox genes in plant development. Annu. Rev. Cell Dev. Biol. 2004, 20, 125–151. [Google Scholar] [CrossRef] [PubMed]
- Hay, A.; Tsiantis, M. A KNOX family tale. Curr. Opin. Plant Biol. 2009, 12, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, K.; Nishiyama, T.; Deguchi, H.; Hasebe, M. Class 1 KNOX genes are not involved in shoot development in the moss Physcomitrella patens but do function in sporophyte development. Evol. Dev. 2008, 10, 555–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Zhang, Y.; Liu, W.; Wang, H.; Wen, S.; Zhang, Y.; Xu, L. Molecular evolution of auxin-mediated root initiation in plants. Mol. Biol. Evol. 2020, 37, 1387–1393. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Yang, X.; Zhao, W.; Lang, T.; Samuelsson, T. Evolution, diversification, and expression of KNOX proteins in plants. Front. Plant Sci. 2015, 6, 882. [Google Scholar] [CrossRef] [Green Version]
- Simpson, M.G. Plant Systematics; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Kenrick, P.; Crane, P.R. The origin and early evolution of plants on land. Nature 1997, 389, 33–39. [Google Scholar] [CrossRef]
- Shen, H.; Jin, D.; Shu, J.P.; Zhou, X.-L.; Lei, M.; Wei, R.; Shang, H.; Wei, H.J.; Zhang, R.; Liu, L. Large-scale phylogenomic analysis resolves a backbone phylogeny in ferns. GigaScience 2018, 7, gix116. [Google Scholar] [CrossRef] [Green Version]
- Schuettpelz, E. The Evolution and Diversification of Epiphytic Ferns; Duke University: Durham, NC, USA, 2007. [Google Scholar]
- Adibah, M.R.; Ainuddin, A. Epiphytic plants responses to light and water stress. Asian J. Plant Sci. 2011, 10, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Bartels, S.F.; Chen, H.Y. Mechanisms regulating epiphytic plant diversity. CRC Crit. Rev. Plant Sci. 2012, 31, 391–400. [Google Scholar] [CrossRef]
- Miller, G.; Hartzell, S.; Porporato, A. Ecohydrology of epiphytes: Modelling water balance, CAM photosynthesis, and their climate impacts. Ecohydrology 2021, 14, e2275. [Google Scholar] [CrossRef]
- Lai, X.; Chahtane, H.; Martin-Arevalillo, R.; Zubieta, C.; Parcy, F. Contrasted evolutionary trajectories of plant transcription factors. Curr. Opin. Plant Biol. 2020, 54, 101–107. [Google Scholar] [CrossRef]
- Debernardi, J.M.; Mecchia, M.A.; Vercruyssen, L.; Smaczniak, C.; Kaufmann, K.; Inze, D.; Rodriguez, R.E.; Palatnik, J.F. Post-transcriptional control of GRF transcription factors by micro RNA miR396 and GIF co-activator affects leaf size and longevity. Plant J. 2014, 79, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Choi, D.; Kende, H. The AtGRF family of putative transcription factors is involved in leaf and cotyledon growth in Arabidopsis. Plant J. 2003, 36, 94–104. [Google Scholar] [CrossRef]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-box gene family in rice: Genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genom. 2007, 8, 242. [Google Scholar] [CrossRef] [Green Version]
- Parenicová, L.; de Folter, S.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: New openings to the MADS world. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef] [Green Version]
- Tapia-López, R.; García-Ponce, B.; Dubrovsky, J.G.; Garay-Arroyo, A.; Pérez-Ruíz, R.V.; Kim, S.-H.; Acevedo, F.; Pelaz, S.; Alvarez-Buylla, E.R. An AGAMOUS-related MADS-box gene, XAL1 (AGL12), regulates root meristem cell proliferation and flowering transition in Arabidopsis. Plant Physiol. 2008, 146, 1182–1192. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.-Q.; Liu, K.-W.; Li, Z.; Lohaus, R.; Hsiao, Y.-Y.; Niu, S.-C.; Wang, J.-Y.; Lin, Y.-C.; Xu, Q.; Chen, L.-J.; et al. The Apostasia genome and the evolution of orchids. Nature 2017, 549, 379–383, Erratum in Nature 2020, 583, E30. [Google Scholar] [CrossRef] [Green Version]
- Van der Graaff, E.; Laux, T.; Rensing, S.A. The WUS homeobox-containing (WOX) protein family. Genome Biol. 2009, 10, 248. [Google Scholar] [CrossRef]
- Samac, D.A.; Litterer, L.; Temple, G.; Jung, H.-J.G.; Somers, D.A. Expression of UDP-glucose dehydrogenase reduces cell-wall polysaccharide concentration and increases xylose content in alfalfa stems. Appl. Biochem. Biotechnol. 2004, 116, 1167–1182. [Google Scholar] [CrossRef]
- Klinghammer, M.; Tenhaken, R. Genome-wide analysis of the UDP-glucose dehydrogenase gene family in Arabidopsis, a key enzyme for matrix polysaccharides in cell walls. J. Exp. Bot. 2007, 58, 3609–3621. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Qi, X.; Chen, D.; Qi, J.; Ma, H. Recurrent genome duplication events likely contributed to both the ancient and recent rise of ferns. J. Integr. Plant Biol. 2020, 62, 433–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Li, W.Z.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Yang, Z. PAML: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Rapidly Evolved TF/TR Families |
|---|---|
| Physcomitrium patens | null |
| Selaginella moellendorffii | null |
| Arabidopsis thaliana | MADS-MIKC [+25 *] |
| Equisetum arvense | AUX-IAA [+14 *], HB-BELL [+12 *], SBP [+16 *], GATA-Tify [+17 *] |
| Angiopteris fokiensis | BES1 [+8 *], HD-ZIP [+16 *], Jumonji [+12 *], MADS-MIKC [+28 *], TRAF [+25 *] |
| Tmesipteris tannensis | Dof [+7 *], GATA-Tify [+9 *], BES1 [−4 *], GRF [−3 *] |
| Psilotum nudum | GRF [−5 *], Dof [−8 *], MADS-MIKC [−5 *] |
| Botrychium japonicum | BES1 [+6 *], GARP-G2-like [+15 *], HB-other [+11 *], IWS1 [+10 *], MYB-related [+17 *], TRAF [+17 *] |
| Ophioglossum vulgatum | null |
| Ophioglossum pendulum | GRF [−3 *], HB-other [−6 *], MADS-MIKC [−5 *] |
| Dipteris chinensis | Dof [−15 *], GARP-G2-like [−17 *], HB-HD-ZIP [−14 *], Jumonji [−11 *], MYB-related [−22 *], SNF2 [−20 *], TRAF [−14 *] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, Z.; Liu, L.; Wei, Z.; Wang, F.; Shen, H.; Yan, Y. Analysis of Comparative Transcriptome and Positively Selected Genes Reveal Adaptive Evolution in Leaf-Less and Root-Less Whisk Ferns. Plants 2022, 11, 1198. https://doi.org/10.3390/plants11091198
Xia Z, Liu L, Wei Z, Wang F, Shen H, Yan Y. Analysis of Comparative Transcriptome and Positively Selected Genes Reveal Adaptive Evolution in Leaf-Less and Root-Less Whisk Ferns. Plants. 2022; 11(9):1198. https://doi.org/10.3390/plants11091198
Chicago/Turabian StyleXia, Zengqiang, Li Liu, Zuoying Wei, Faguo Wang, Hui Shen, and Yuehong Yan. 2022. "Analysis of Comparative Transcriptome and Positively Selected Genes Reveal Adaptive Evolution in Leaf-Less and Root-Less Whisk Ferns" Plants 11, no. 9: 1198. https://doi.org/10.3390/plants11091198
APA StyleXia, Z., Liu, L., Wei, Z., Wang, F., Shen, H., & Yan, Y. (2022). Analysis of Comparative Transcriptome and Positively Selected Genes Reveal Adaptive Evolution in Leaf-Less and Root-Less Whisk Ferns. Plants, 11(9), 1198. https://doi.org/10.3390/plants11091198