
Stochastic Variation in DNA Methylation Modulates Nucleosome Occupancy and Alternative Splicing in Arabidopsis thaliana


 ,
,  ,
,  and
and 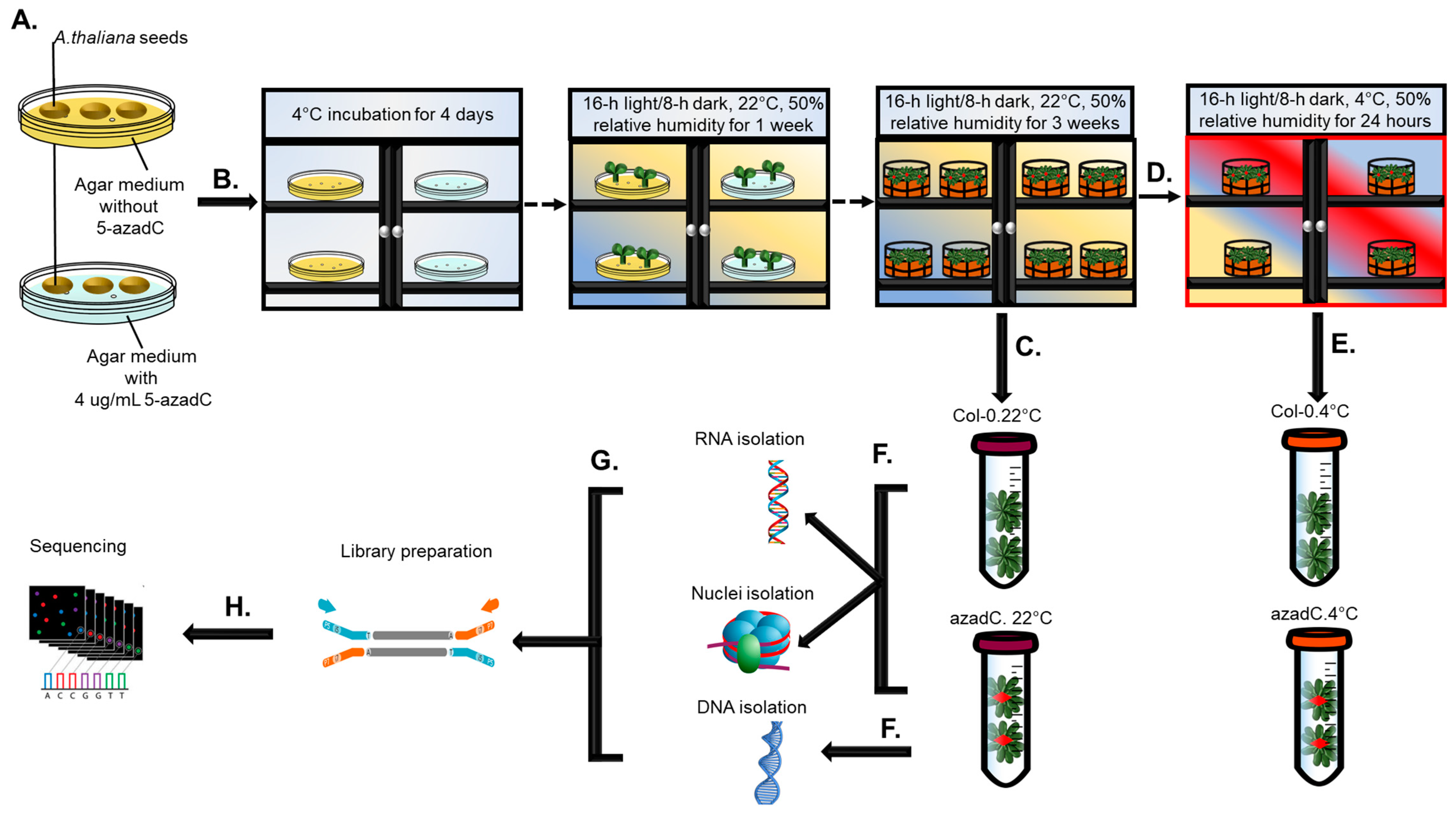{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
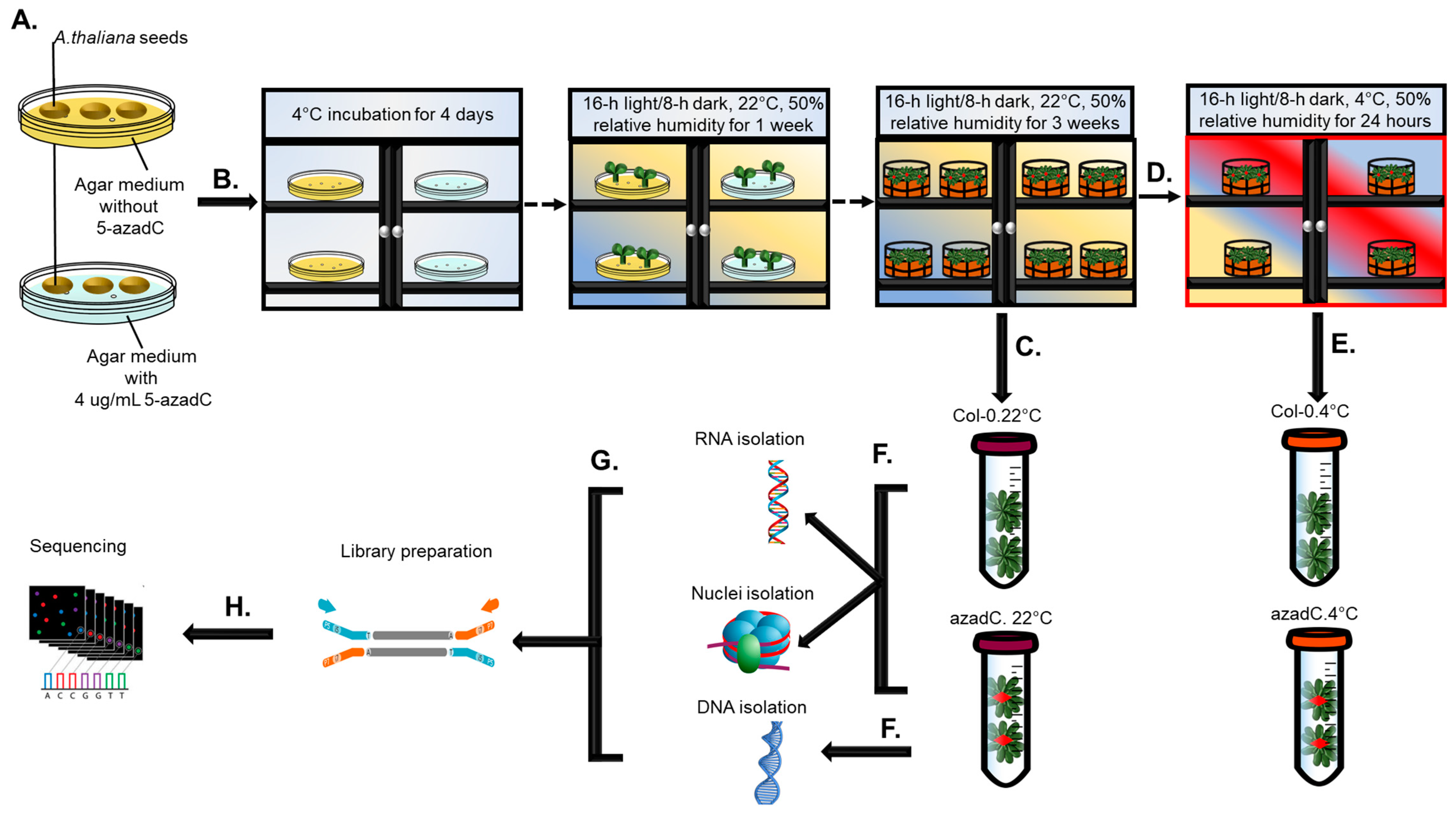
2.1. Plant Growth and Chemical Treatments
2.2. RNA and DNA Extraction
2.3. Nuclei Isolation and Digestion with MNase
2.4. Library Preparation and Sequencing Information
2.5. DE and DAS Analysis
2.6. Parametric Gene Set Enrichment Analysis
2.7. Gene Ontology Analysis
2.8. Identification of AS Events, PSI, and Delta PSI from RNA-Seq Data
2.9. MNase-Seq Data Processing
2.10. Differential Nucleosome Positioning
2.11. Bisulphite-Sequencing Data Processing
3. Results
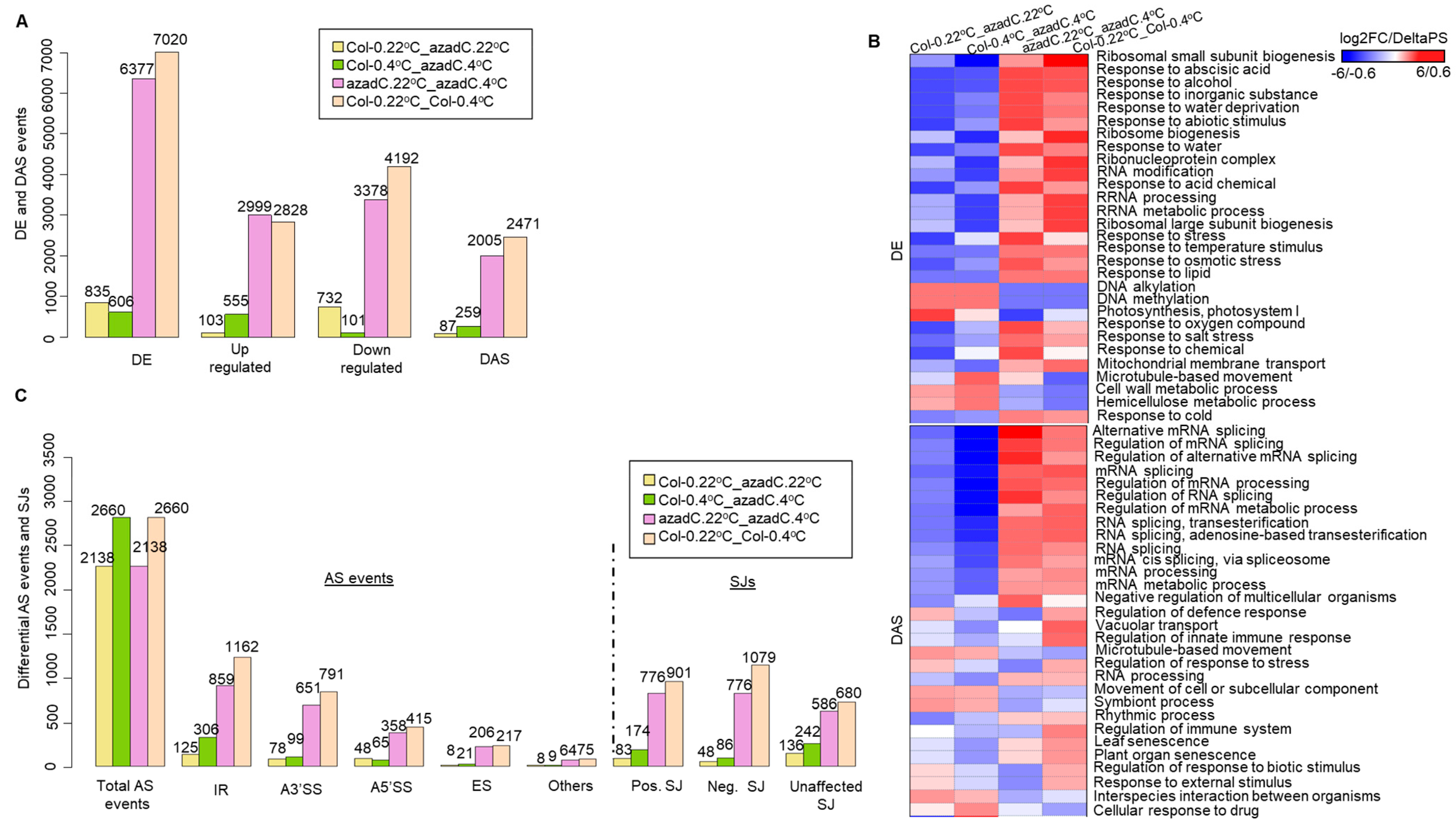
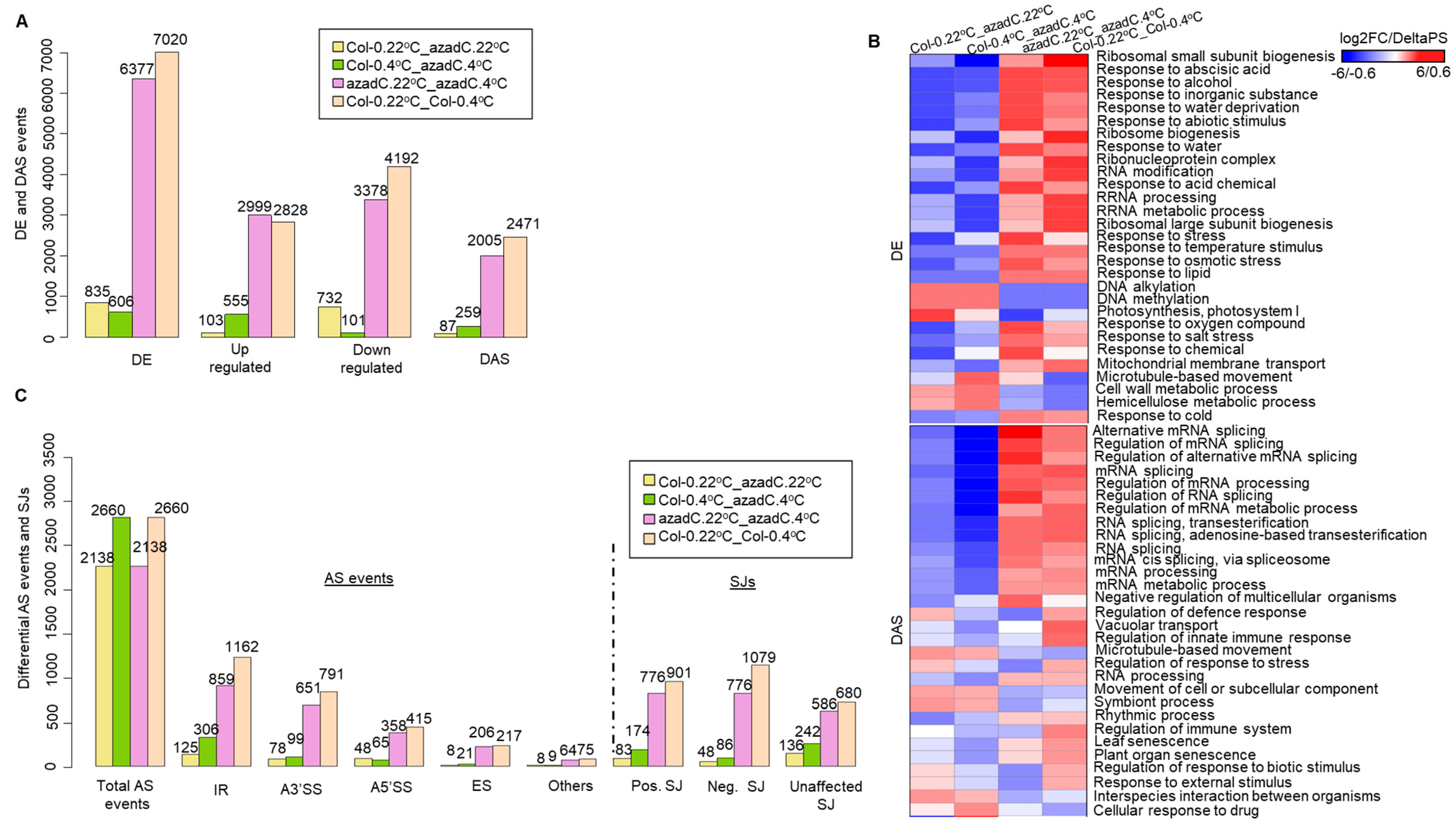
3.1. 5-Aza-2′-Deoxycytidine Treatment Affects Gene Expression and Alternative Splicing Profiles
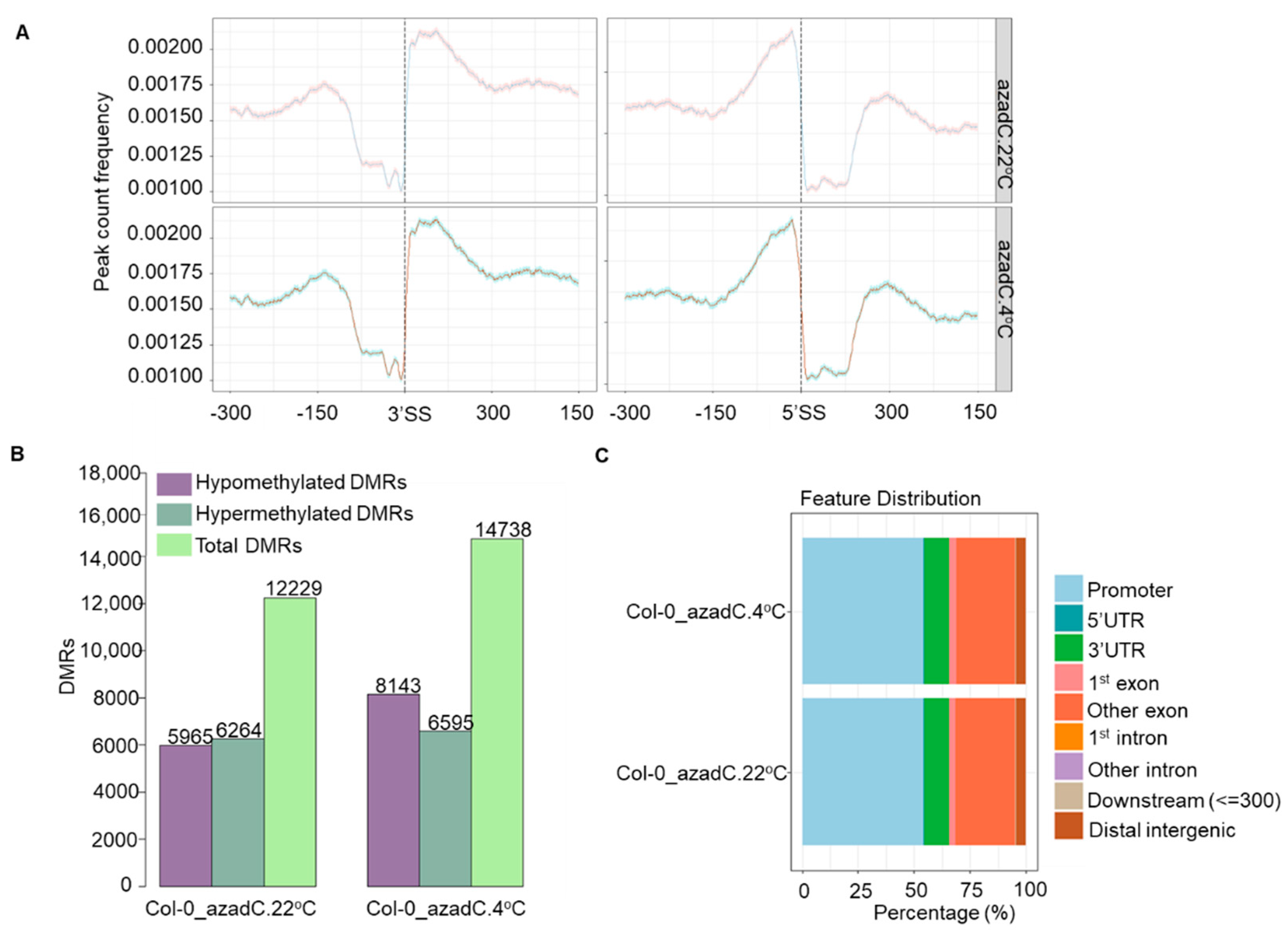
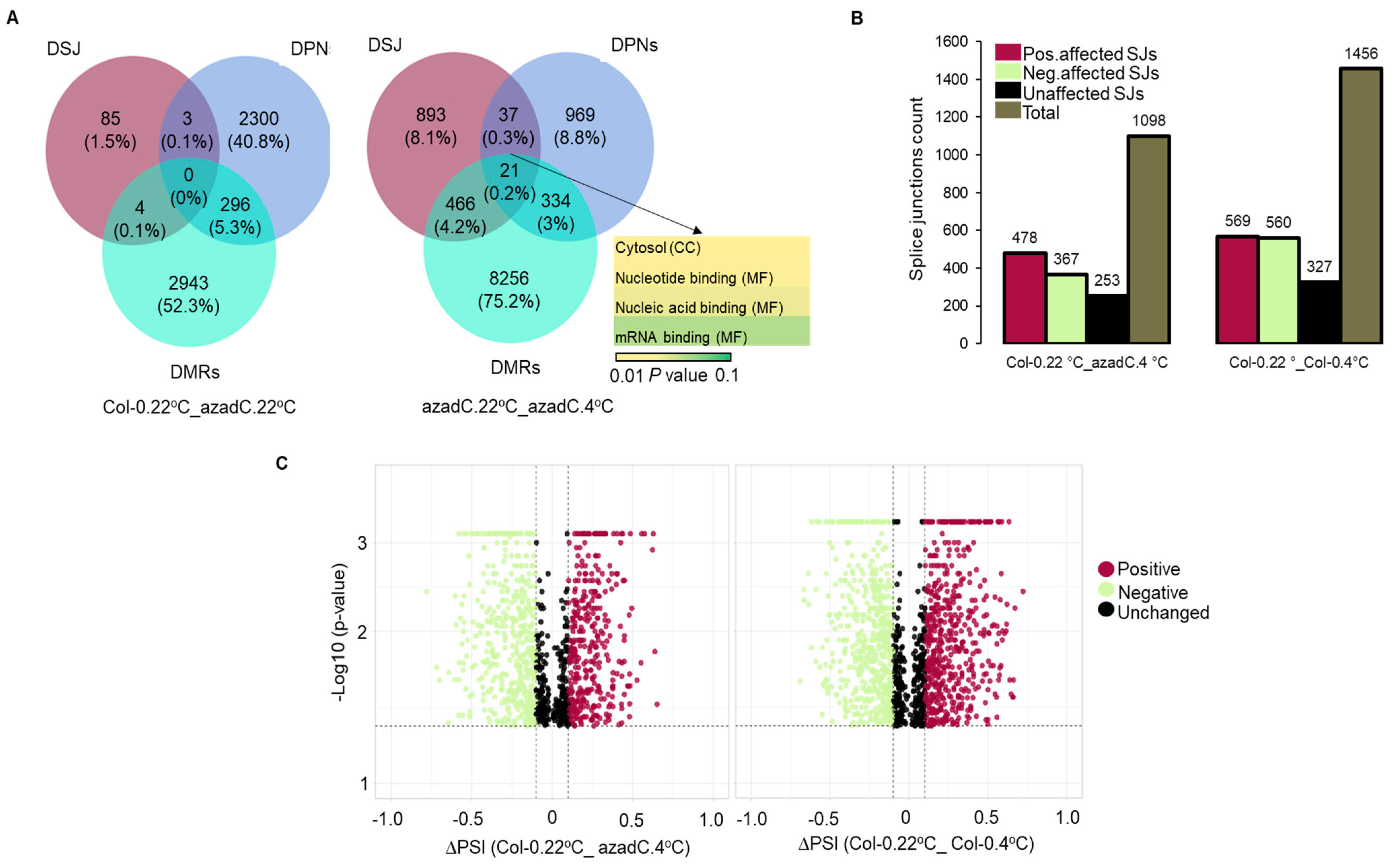
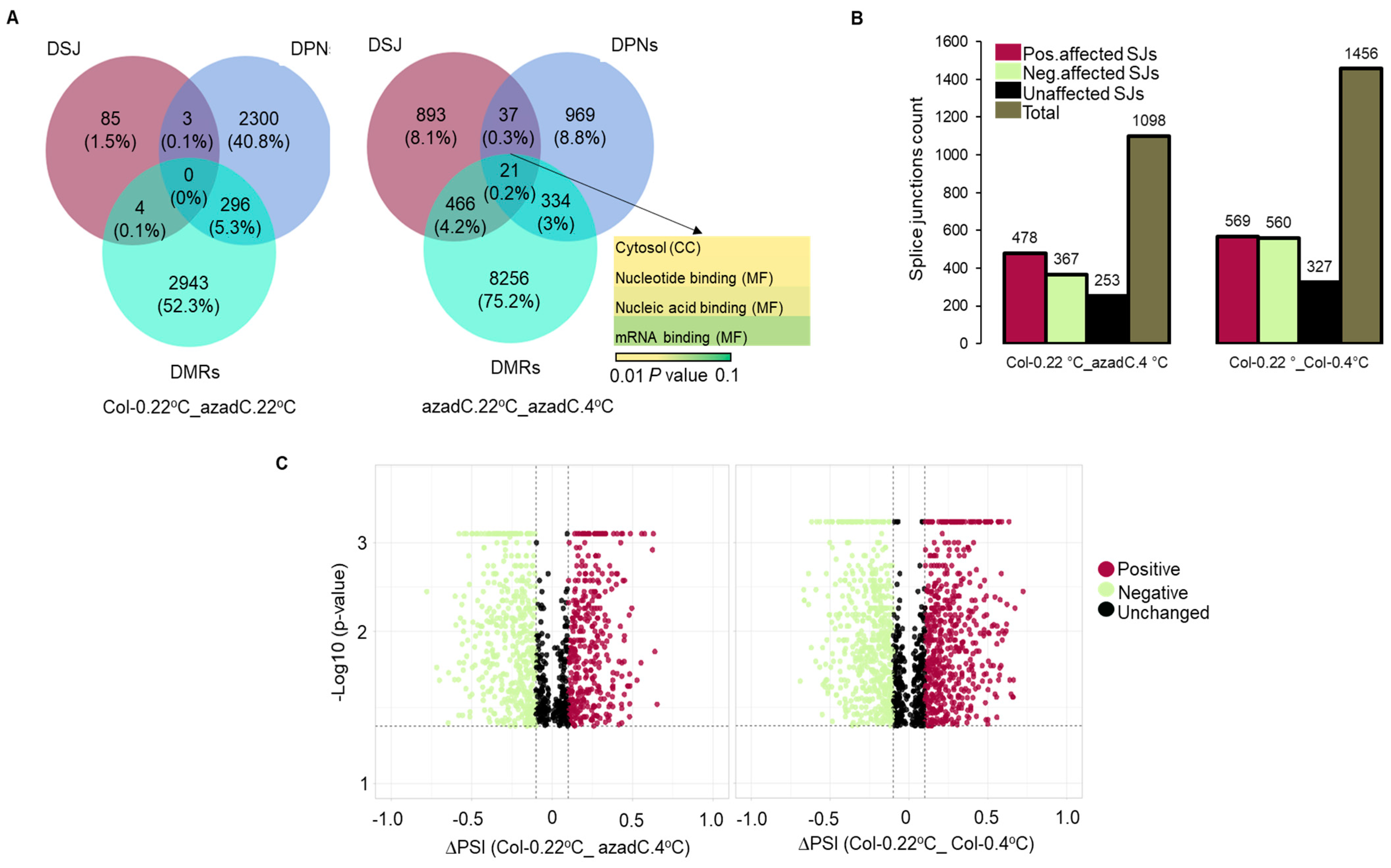
3.2. Stochastic DNA Methylation Affects Splice Junctions and AS Event Profiles
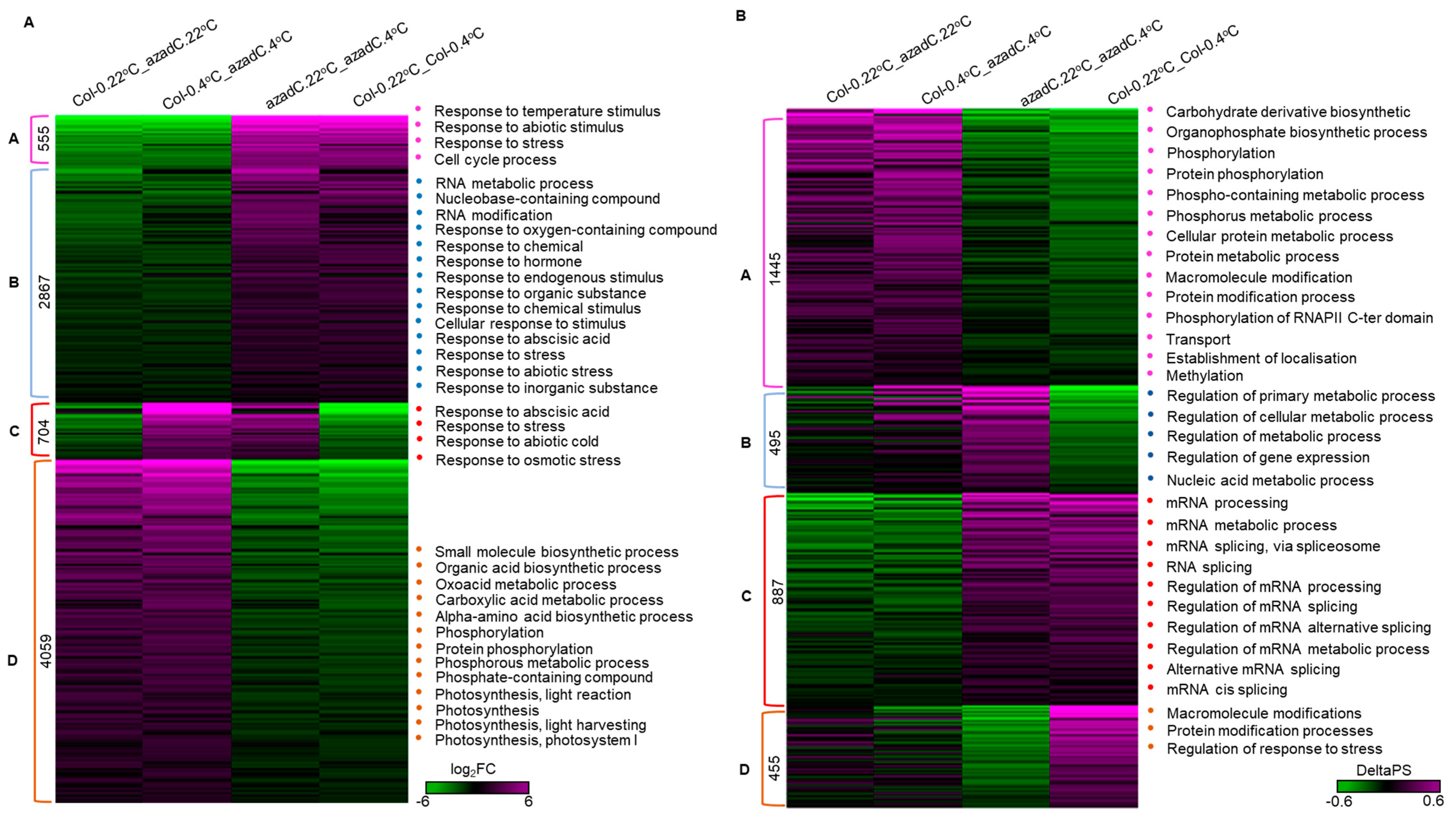
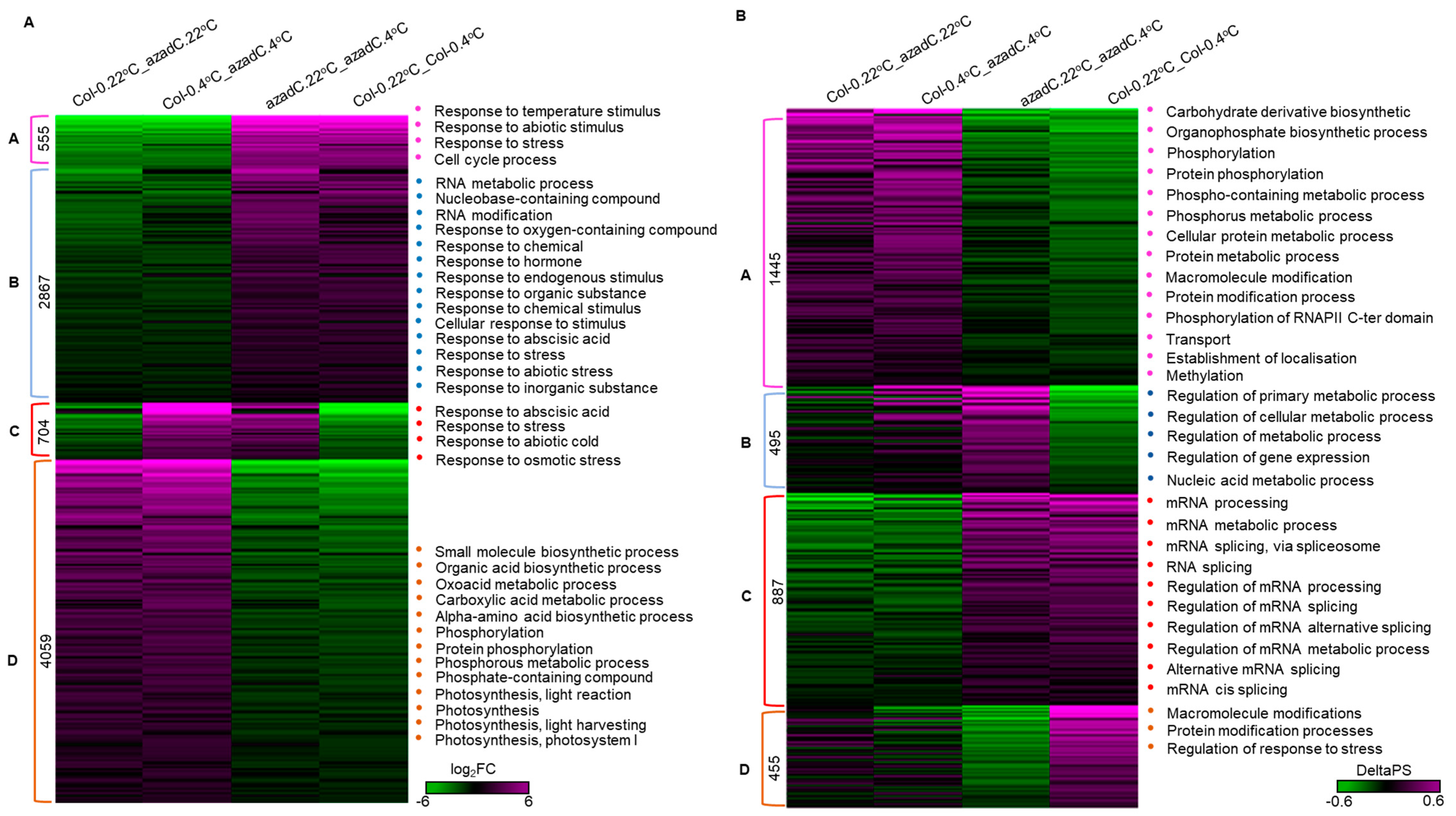
3.3. DNA Methylation Fine-Tunes Gene Expression
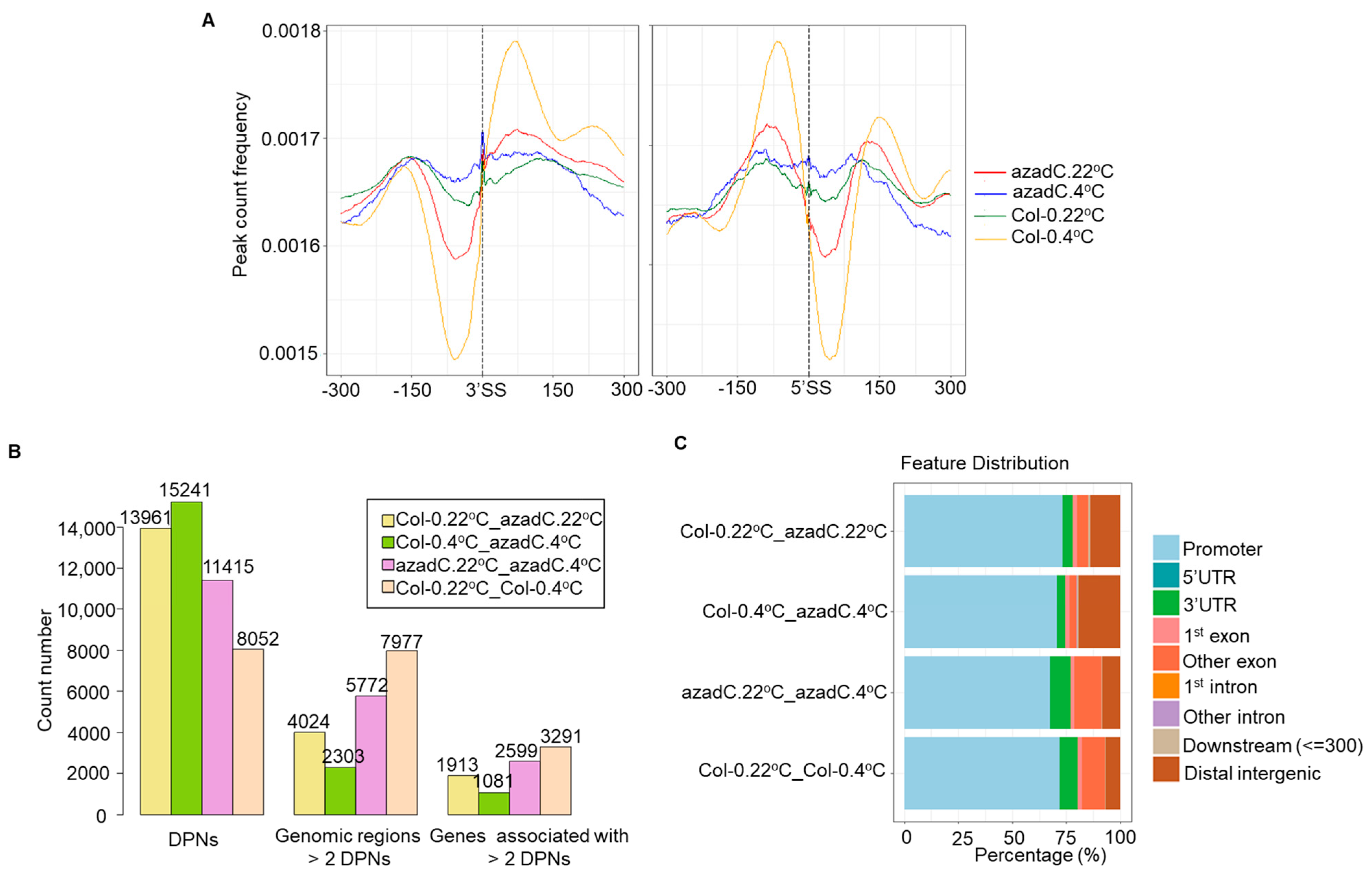
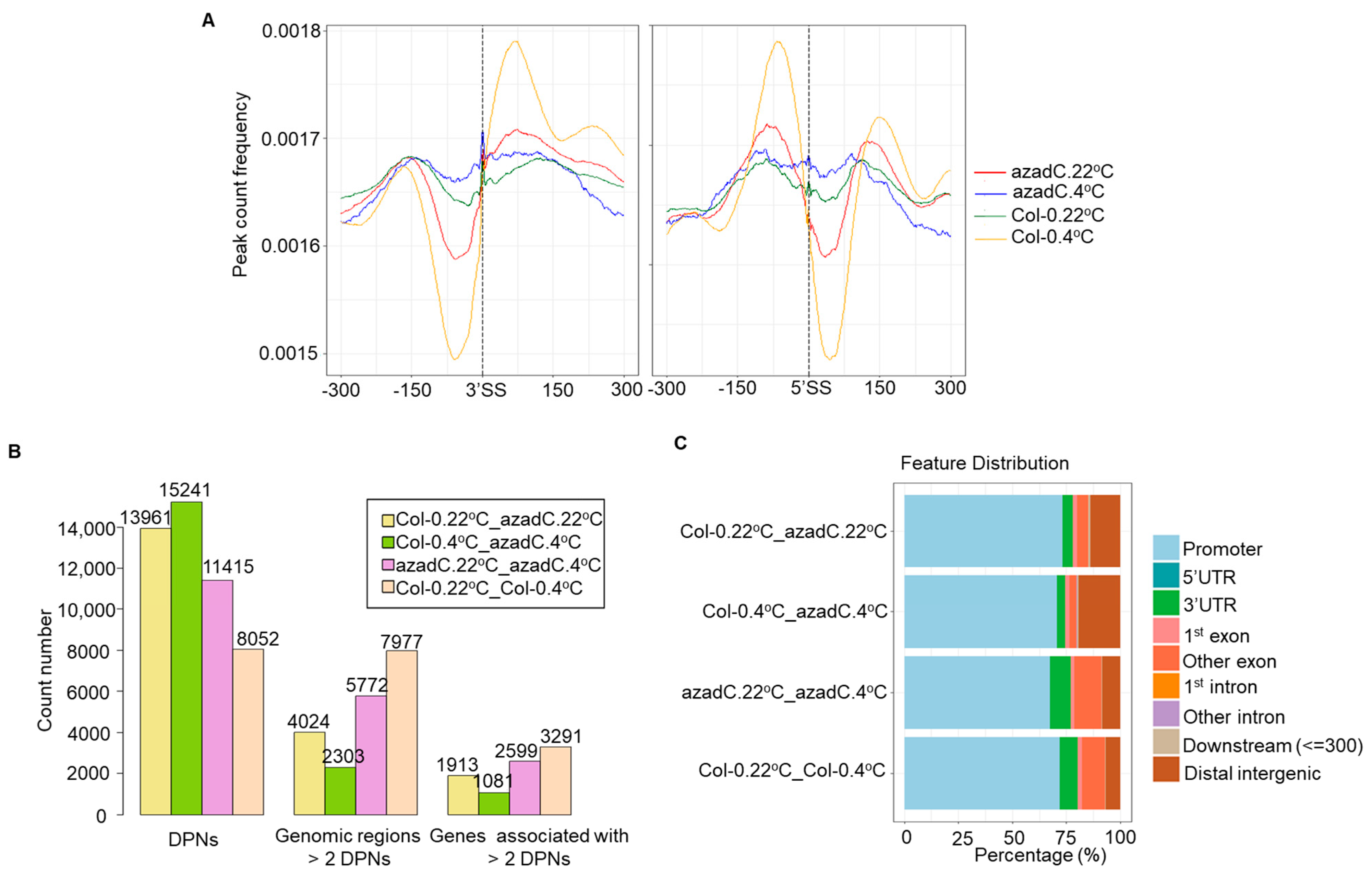
3.4. DNA Methylation Levels Modulate Nucleosome Occupancy in Response to Cold Stress
3.5. DNA-Dependant Co-Transcriptional Regulation of Key Metabolic Process under Cold Stress
3.6. Epigenetic Features Regulate the Directionality of Splicing upon Cold Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, H.; Tian, C.; Yu, Y.; Jiao, Y. Transcriptome Survey of the Contribution of Alternative Splicing to Proteome Diversity in Arabidopsis thaliana. Mol. Plant 2016, 9, 749–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Feng, L.; Li, J.; He, Z. Genetic and epigenetic control of plant heat responses. Front. Plant Sci. 2015, 6, 267. [Google Scholar] [CrossRef] [PubMed]
- Syed, N.H.; Kalyna, M.; Marquez, Y.; Barta, A.; Brown, J.W.S. Alternative splicing in plants—Coming of age. Trends Plant Sci. 2012, 17, 616–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, A.S.N.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabre, I.; Reddy, A.S.N.; Kalyna, M.; Chaudhary, S.; Khokhar, W.; Byrne, L.J.; Wilson, C.M.; Syed, N.H. Does co-transcriptional regulation of alternative splicing mediate plant stress responses? Nucleic Acids Res. 2019, 47, 2716–2726. [Google Scholar] [CrossRef] [PubMed]
- Marquez, Y.; Brown, J.W.S.; Simpson, C.; Barta, A.; Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012, 22, 1184–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Guo, G.; Hu, X.; Zhang, Y.; Li, Q.; Li, R.; Zhuang, R.; Lu, Z.; He, Z.; Fang, X.; et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20, 646–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamala, S.; Feng, G.; Chavarro, C.; Barbazuk, W.B. Genome-Wide Identification of Evolutionarily Conserved Alternative Splicing Events in Flowering Plants. Front. Bioeng. Biotechnol. 2015, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filichkin, S.A.; Hamilton, M.; Dharmawardhana, P.D.; Singh, S.K.; Sullivan, C.; Ben-Hur, A.; Reddy, A.S.; Jaiswal, P. Abiotic stresses modulate landscape of poplar transcriptome via alternative splicing, differential intron retention, and isoform ratio switching. Front. Plant Sci. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrangelo, A.M.; Marone, D.; Laidò, G.; De Leonardis, A.M.; De Vita, P. Alternative splicing: Enhancing ability to cope with stress via transcriptome plasticity. Plant Sci. 2012, 185–186, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.; Jabre, I.; Reddy, A.S.N.; Staiger, D.; Syed, N.H. Perspective on Alternative Splicing and Proteome Complexity in Plants. Trends Plant Sci. 2019, 24, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Calixto, C.P.G.; Guo, W.; James, A.B.; Tzioutziou, N.A.; Entizne, J.C.; Panter, P.E.; Knight, H.; Nimmo, H.; Zhang, R.; Brown, J.W.S. Rapid and dynamic alternative splicing impacts the Arabidopsis cold response transcriptome. Plant Cell 2018, 30, 1424–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streitner, C.; Simpson, C.G.; Shaw, P.; Danisman, S.; Brown, J.W.S.; Staiger, D. Small changes in ambient temperature affect alternative splicing in Arabidopsis thaliana. Plant Signal. Behav. 2013, 8, e24638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, A.B.; Syed, N.H.; Bordage, S.; Marshall, J.; Nimmo, G.A.; Jenkins, G.I.; Herzyk, P.; Brown, J.W.S.; Nimmo, H.G. Alternative Splicing Mediates Responses of the Arabidopsis Circadian Clock to Temperature Changes. Plant Cell 2012, 24, 961–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filichkin, S.A.; Cumbie, J.S.; Dharmawardhana, P.; Jaiswal, P.; Chang, J.H.; Palusa, S.G.; Reddy, A.S.N.; Megraw, M.; Mockler, T.C. Environmental stresses modulate abundance and timing of alternatively spliced circadian transcripts in Arabidopsis. Mol. Plant 2015, 8, 207–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Tang, Y.; Kwok, C.K.; Zhang, Y.; Bevilacqua, P.C.; Assmann, S.M. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2014, 505, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Kan, J.L.C.; Green, M.R. Arginine-Serine-Rich Domains Bound at Splicing Enhancers Contact the Branchpoint to Promote Prespliceosome Assembly. Mol. Cell 2004, 13, 367–376. [Google Scholar] [CrossRef]
- Hajheidari, M.; Koncz, C.; Eick, D. Emerging roles for RNA polymerase II CTD in Arabidopsis. Trends Plant Sci. 2013, 18, 633–643. [Google Scholar] [CrossRef]
- Lenasi, T.; Barboric, M. P-TEFb stimulates transcription elongation and pre-mRNA splicing through multilateral mechanisms. RNA Biol. 2010, 7, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Hirose, Y.; Manley, J.L. RNA polymerase II and the integration of nuclear events. Genes Dev. 2000, 14, 1415–1429. [Google Scholar] [CrossRef]
- Gasch, A.; Wiesner, S.; Martin-Malpartida, P.; Ramirez-Espain, X.; Ruiz, L.; Macias, M.J. The structure of Prp40 FF1 domain and its interaction with the crn-TPR1 motif of Clf1 gives a new insight into the binding mode of FF domains. J. Biol. Chem. 2006, 281, 356–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, S.; Jabre, I.; Syed, N.H. Epigenetic differences in an identical genetic background modulate alternative splicing in A. thaliana. Genomics 2021, 113, 3476–3486. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Gama-Sosa, M.A.; Huang, L.H.; Midgett, R.M.; Kuo, K.C.; Mccune, R.A.; Gehrke, C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues or cells. Nucleic Acids Res. 1982, 10, 2709–2721. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chodavarapu, R.K.; Feng, S.; Bernatavichute, Y.V.; Chen, P.-Y.Y.; Stroud, H.; Yu, Y.; Hetzel, J.A.; Kuo, F.; Kim, J.; Cokus, S.J.; et al. Relationship between nucleosome positioning and DNA methylation. Nature 2010, 466, 388–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, S.; Meshorer, E.; Ast, G. Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 2009, 16, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Mavrich, T.N.; Jiang, C.; Ioshikhes, I.P.; Li, X.; Venters, B.J.; Zanton, S.J.; Tomsho, L.P.; Qi, J.; Glaser, R.L.; Schuster, S.C.; et al. Nucleosome organization in the Drosophila genome. Nature 2008, 453, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-J.; Seddon, A.E.; Tsai, Z.T.-Y.; Major, I.T.; Floer, M.; Howe, G.A.; Shiu, S.-H. Determinants of nucleosome positioning and their influence on plant gene expression. Genome Res. 2015, 25, 1182–1195. [Google Scholar] [CrossRef]
- Gelfman, S.; Cohen, N.; Yearim, A.; Ast, G. DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon-intron structure. Genome Res. 2013, 23, 789–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilgner, H.; Nikolaou, C.; Althammer, S.; Sammeth, M.; Beato, M.; Valcárcel, J.; Guigó, R. Nucleosome positioning as a determinant of exon recognition. Nat. Struct. Mol. Biol. 2009, 16, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Luo, L.; Zhang, L. The organization of nucleosomes around splice sites. Nucleic Acids Res. 2010, 38, 2788–2798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahkuri, S.; Taft, R.J.; Mattick, J.S. Nucleosomes are preferentially positioned at exons in somatic and sperm cells. Cell Cycle 2009, 8, 3420–3424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huff, J.T.; Zilberman, D. Dnmt1-independent CG methylation contributes to nucleosome positioning in diverse eukaryotes. Cell 2014, 156, 1286–1297. [Google Scholar] [CrossRef] [Green Version]
- Alexander, R.D.; Innocente, S.A.; Barrass, J.D.; Beggs, J.D. Splicing-Dependent RNA polymerase pausing in yeast. Mol. Cell 2010, 40, 582–593. [Google Scholar] [CrossRef]
- Ullah, F.; Hamilton, M.; Reddy, A.S.N.; Ben-Hur, A. Exploring the relationship between intron retention and chromatin accessibility in plants. BMC Genom. 2018, 19, 21. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Liu, M.; Liu, X.; Dong, Z. RNA polymerase II activity revealed by GRO-seq and pNET-seq in Arabidopsis. Nat. Plants 2018, 4, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Narayana Chevala, V.; Shankar, R.; Jain, M. Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci. Rep. 2015, 5, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secco, D.; Wang, C.; Shou, H.; Schultz, M.D.; Chiarenza, S.; Nussaume, L.; Ecker, J.R.; Whelan, J.; Lister, R. Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. Elife 2015, 4, e.09343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Wang, X.; Chen, X.; Shu, N.; Wang, J.; Wang, D.; Wang, S.; Fan, W.; Guo, L.; Guo, X.; et al. Single-base resolution methylomes of upland cotton (Gossypium hirsutum L.) reveal epigenome modifications in response to drought stress. BMC Genom. 2017, 18, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steward, N.; Ito, M.; Yamaguchi, Y.; Koizumi, N.; Sano, H. Periodic DNA methylation in maize nucleosomes and demethylation by environmental stress. J. Biol. Chem. 2002, 277, 37741–37746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef] [PubMed]
- Christman, J. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, H.; Kamada, I.; Youssefian, S.; Katsumi, M.; Wabiko, H. A single treatment of rice seedlings with 5-azacytidine induces heritable dwarfism and undermethylation of genomic DNA. MGG Mol. Gen. Genet. 1990, 220, 441–447. [Google Scholar] [CrossRef]
- Kumpatla, S.P.; Teng, W.; Buchholz, W.G.; Hall, T.C. Epigenetic Transcriptional Silencing and 5-Azacytidine-Mediated Reactivation of a Complex Transgene in Rice. Plant Physiol. 1997, 115, 361–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fieldes, M.A. Heritable effects of 5-azacytidine treatments on the growth and development of flax (Linum usitatissimum) genotrophs and genotypes. Genome 1994, 37, 1–11. [Google Scholar] [CrossRef]
- Vyskot, B.; Koukalová, B.; Kovařík, A.; Sachambula, L.; Reynolds, D.; Bezděk, M. Meiotic transmission of a hypomethylated repetitive DNA family in tobacco. Theor. Appl. Genet. 1995, 91, 659–664. [Google Scholar] [CrossRef] [PubMed]
- King, G.J. Morphological development in Brassica oleracea is modulated by in vivo treatment with 5-azacytidine. J. Hortic. Sci. 1995, 70, 333–342. [Google Scholar] [CrossRef]
- Janoušek, B.; Široký, J.; Vyskot, B. Epigenetic control of sexual phenotype in a dioecious plant, Melandrium album. Mol. Gen. Genet. 1996, 250, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Amado, L.; Abranches, R.; Neves, N.; Viegas, W. Development-dependent inheritance of 5-azacytidine-induced epimutations in triticale: Analysis of rDNA expression patterns. Chromosom. Res. 1997, 5, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Burn, J.E.; Bagnall, D.J.; Metzger, J.D.; Dennis, E.S.; Peacock, W.J. DNA methylation, vernalization, and the initiation of flowering. Proc. Natl. Acad. Sci. USA 1993, 90, 287–291. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Tanino, K.K.; Horner, K.N.; Robinson, S.J. Quantitative trait variation is revealed in a novel hypomethylated population of woodland strawberry (Fragaria vesca). BMC Plant Biol. 2016, 16, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marfil, C.F.; Asurmendi, S.; Masuelli, R.W. Changes in micro RNA expression in a wild tuber-bearing Solanum species induced by 5-Azacytidine treatment. Plant Cell Rep. 2012, 31, 1449–1461. [Google Scholar] [CrossRef]
- Thomashow, M.F. Molecular Basis of Plant Cold Acclimation: Insights Gained from Studying the CBF Cold Response Pathway. Plant Physiol. 2010, 154, 571–577. [Google Scholar] [CrossRef] [Green Version]
- Knight, M.R.; Knight, H. Low-temperature perception leading to gene expression and cold tolerance in higher plants. New Phytol. 2012, 195, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Barrero-Gil, J.; Salinas, J. Post-translational regulation of cold acclimation response. Plant Sci. 2013, 205–206, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Palusa, S.G.; Ali, G.S.; Reddy, A.S.N. Alternative splicing of pre-mRNAs of Arabidopsis serine/arginine-rich proteins: Regulation by hormones and stresses. Plant J. 2007, 49, 1091–1107. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, W.; Zhao, X.; Zhu, L.; Fu, B.; Li, Z. DNA methylation alterations of rice in response to cold stress. Plant Omics 2011, 4, 364–369. [Google Scholar]
- McClung, C.R.; Davis, S.J. Ambient thermometers in plants: From physiological outputs towards mechanisms of thermal sensing. Curr. Biol. 2010, 20, 1086–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.V.; Wigge, P.A. H2A.Z-Containing Nucleosomes Mediate the Thermosensory Response in Arabidopsis. Cell 2010, 140, 136–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, D.; Paul, A.; Roy, A.; Ghosh, R.; Ganguly, P.; Chaudhuri, S. Differential acetylation of histone H3 at the regulatory region of OsDREB1b promoter facilitates chromatin remodelling and transcription activation during cold stress. PLoS ONE 2014, 9, e100343. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Calixto, C.P.G.; Marquez, Y.; Venhuizen, P.; Tzioutziou, N.A.; Guo, W.; Spensley, M.; Entizne, J.C.; Lewandowska, D.; Have, S.T.; et al. A high quality Arabidopsis transcriptome for accurate transcript-level analysis of alternative splicing. Nucleic Acids Res. 2017, 45, 5061–5073. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Bullard, J.H.; Purdom, E.; Hansen, K.D.; Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinform. 2010, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamancos, G.P.; Pagès, A.; Trincado, J.L.; Bellora, N.; Eyras, E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 2015, 21, 1521–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Liu, Y.; Zhu, S.; Green, C.D.; Wei, G.; Han, J.D.J. Improved nucleosome-positioning algorithm iNPS for accurate nucleosome positioning from sequencing data. Nat. Commun. 2014, 18, 4909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Xi, Y.; Pan, X.; Li, Z.; Kaestner, K.; Tyler, J.; Dent, S.; He, X.; Li, W. DANPOS: Dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 2013, 23, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. MethylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Griffin, P.T.; Niederhuth, C.E.; Schmitz, R.J. A comparative analysis of 5-azacytidine-and zebularine-induced DNA demethylation. G3 Genes Genomes Genet. 2016, 6, 2773–2780. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hu, L.; Wang, X.; Li, N.; Xu, C.; Gong, L.; Liu, B. DNA Methylation Affects Gene Alternative Splicing in Plants: An Example from Rice. Mol. Plant 2016, 9, 305–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Harris, C.J.; Liu, Q.; Liu, W.; Ausin, I.; Long, Y.; Xiao, L.; Feng, L.; Chen, X.; Xie, Y.; et al. Large-scale comparative epigenomics reveals hierarchical regulation of non-CG methylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, E1069–E1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, J.; Jacobsen, S. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wu, T.; Li, S.; He, Q.; Yang, Z.; Zhang, W. The methylation patterns and transcriptional responses to chilling stress at the seedling stage in rice. Int. J. Mol. Sci. 2019, 20, 5089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabre, I.; Chaudhary, S.; Guo, W.; Kalyna, M.; Reddy, A.S.N.; Chen, W.; Zhang, R.; Wilson, C.; Syed, N.H. Differential nucleosome occupancy modulates alternative splicing in Arabidopsis thaliana. New Phytol. 2021, 229, 1937–1945. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chen, L.; Xia, H.; Wei, H.; Lou, Q.; Li, M.; Li, T.; Luo, L. Transgenerational epimutations induced by multi-generation drought imposition mediate rice plant’s adaptation to drought condition. Sci. Rep. 2017, 7, 39843. [Google Scholar] [CrossRef] [PubMed]
- Bäurle, I.; Trindade, I. Chromatin regulation of somatic abiotic stress memory. J. Exp. Bot. 2020, 71, 5269–5279. [Google Scholar] [CrossRef] [Green Version]
- Boyko, A.; Blevins, T.; Yao, Y.; Golubov, A.; Bilichak, A.; Ilnytskyy, Y.; Hollander, J.; Meins, F.; Kovalchuk, I. Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS ONE 2010, 5, e9514. [Google Scholar] [CrossRef]
- Xie, H.; Sun, Y.; Cheng, B.; Xue, S.; Cheng, D.; Liu, L.; Meng, L.; Qiang, S. Variation in ICE1 methylation primarily determines phenotypic variation in freezing tolerance in Arabidopsis thaliana. Plant Cell Physiol. 2019, 60, 152–165. [Google Scholar] [CrossRef]
- Chang, S.; Pikaard, C.S. Transcript profiling in Arabidopsis reveals complex responses to global inhibition of DNA methylation and histone deacetylation. J. Biol. Chem. 2005, 280, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Preite, V.; Oplaat, C.; Biere, A.; Kirschner, J.; Van Der Putten, W.H.; Verhoeven, K.J.F. Increased transgenerational epigenetic variation, but not predictable epigenetic variants, after environmental exposure in two apomictic dandelion lineages. Ecol. Evol. 2018, 8, 3047–3059. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jabre, I.; Chaudhary, S.; Wilson, C.M.; Staiger, D.; Syed, N. Stochastic Variation in DNA Methylation Modulates Nucleosome Occupancy and Alternative Splicing in Arabidopsis thaliana. Plants 2022, 11, 1105. https://doi.org/10.3390/plants11091105
Jabre I, Chaudhary S, Wilson CM, Staiger D, Syed N. Stochastic Variation in DNA Methylation Modulates Nucleosome Occupancy and Alternative Splicing in Arabidopsis thaliana. Plants. 2022; 11(9):1105. https://doi.org/10.3390/plants11091105
Chicago/Turabian StyleJabre, Ibtissam, Saurabh Chaudhary, Cornelia M. Wilson, Dorothee Staiger, and Naeem Syed. 2022. "Stochastic Variation in DNA Methylation Modulates Nucleosome Occupancy and Alternative Splicing in Arabidopsis thaliana" Plants 11, no. 9: 1105. https://doi.org/10.3390/plants11091105
APA StyleJabre, I., Chaudhary, S., Wilson, C. M., Staiger, D., & Syed, N. (2022). Stochastic Variation in DNA Methylation Modulates Nucleosome Occupancy and Alternative Splicing in Arabidopsis thaliana. Plants, 11(9), 1105. https://doi.org/10.3390/plants11091105