
Reevaluation of Parasynechococcus-like Strains and Genomic Analysis of Their Microsatellites and Compound Microsatellites




Abstract
:1. Introduction
2. Results
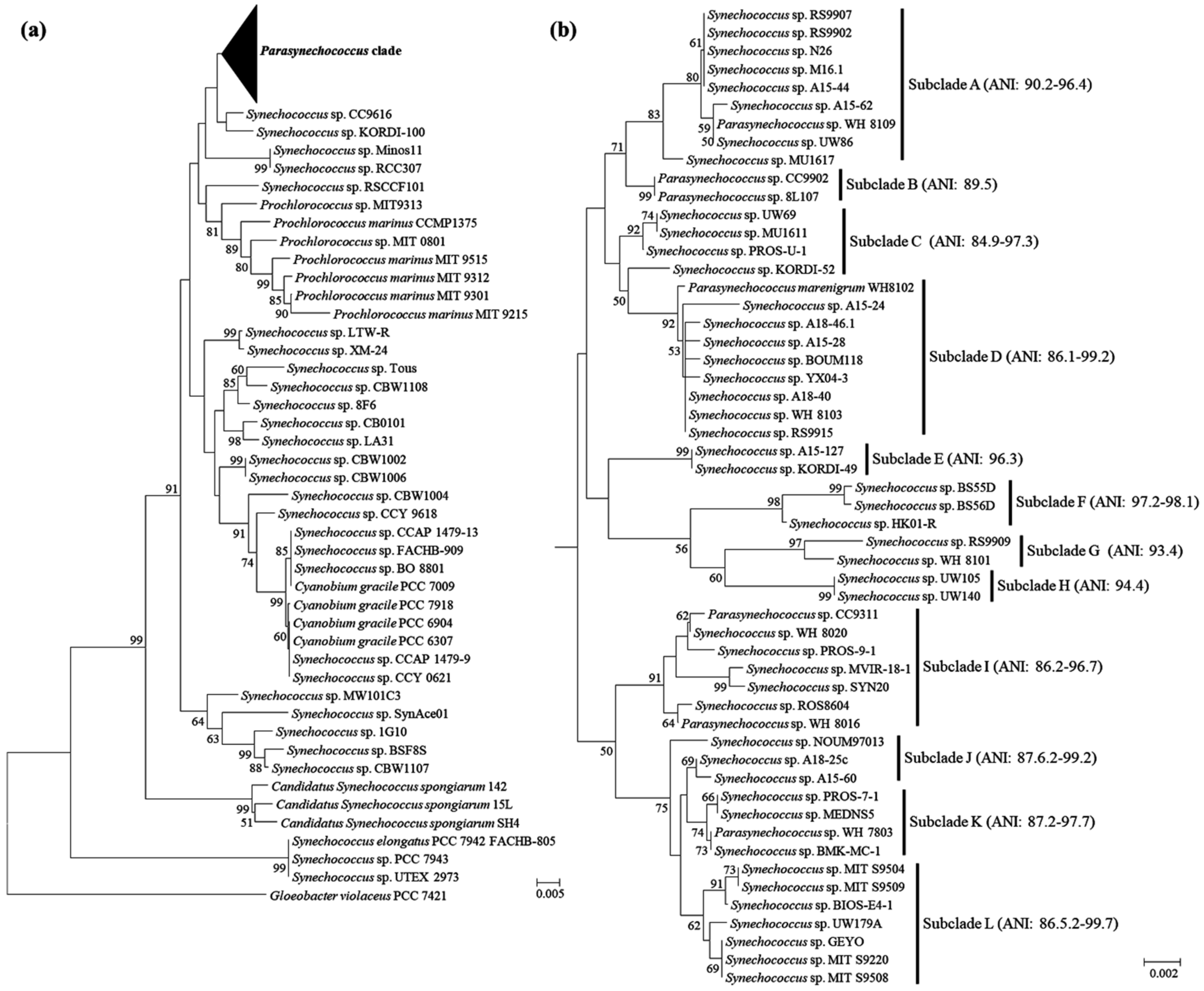
2.1. Reevaluation of Synechococcus-like Strains
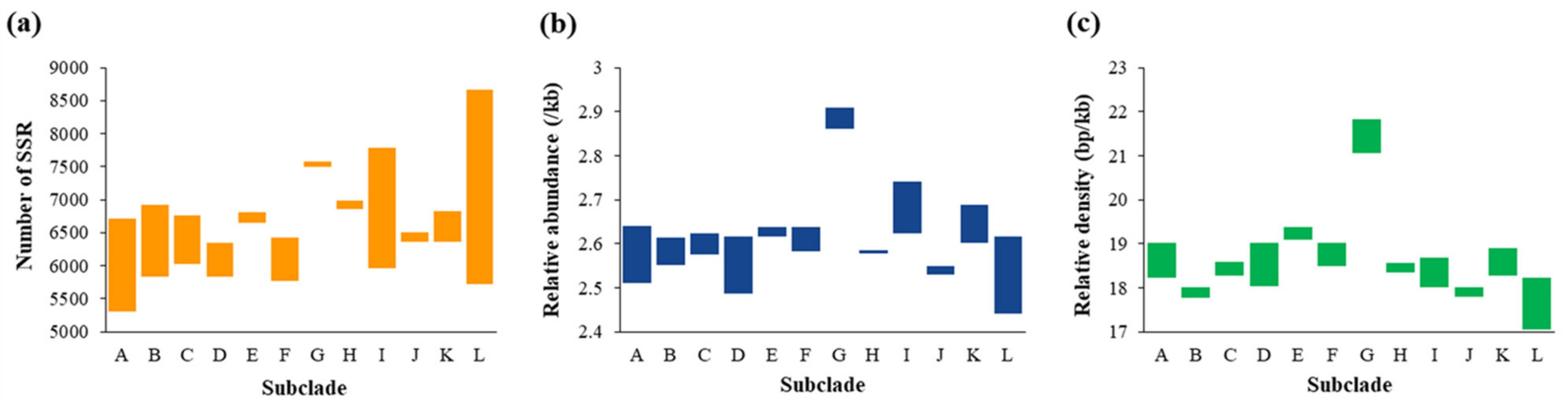
2.2. Number, Relative Abundance (RA), and Relative Density (RD) of SSRs and CSSRs
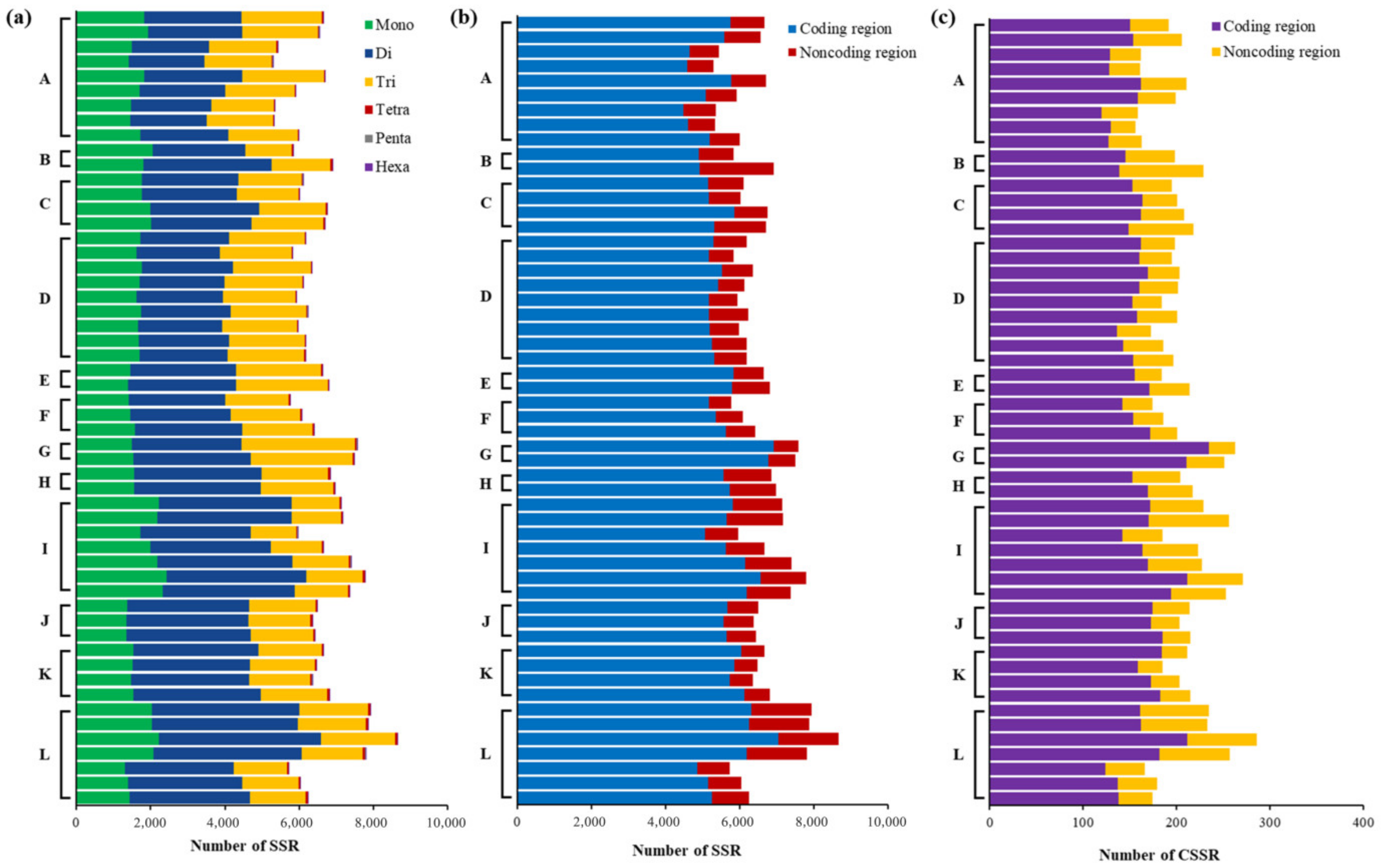
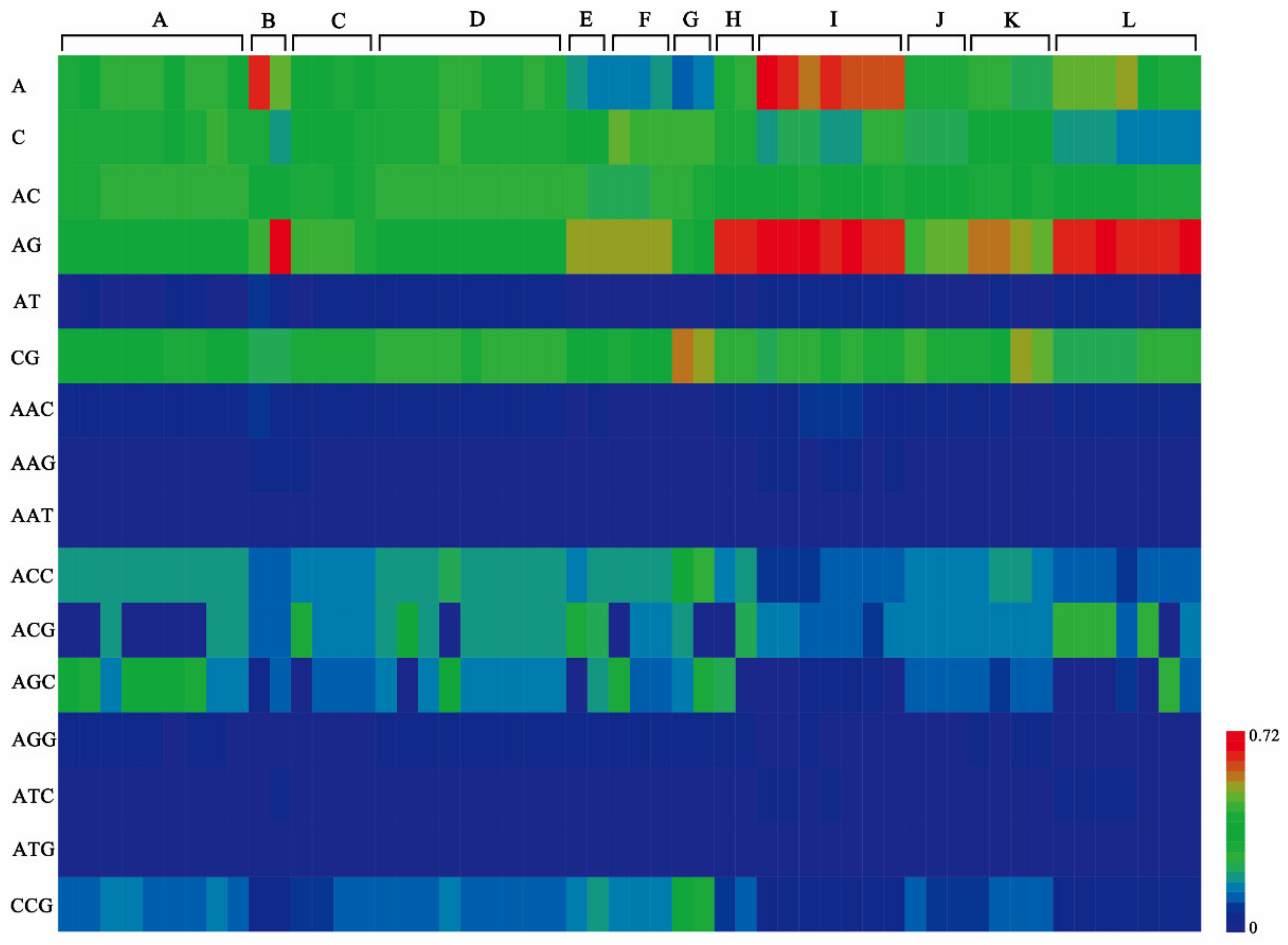
2.3. Distribution and Diversity of SSRs
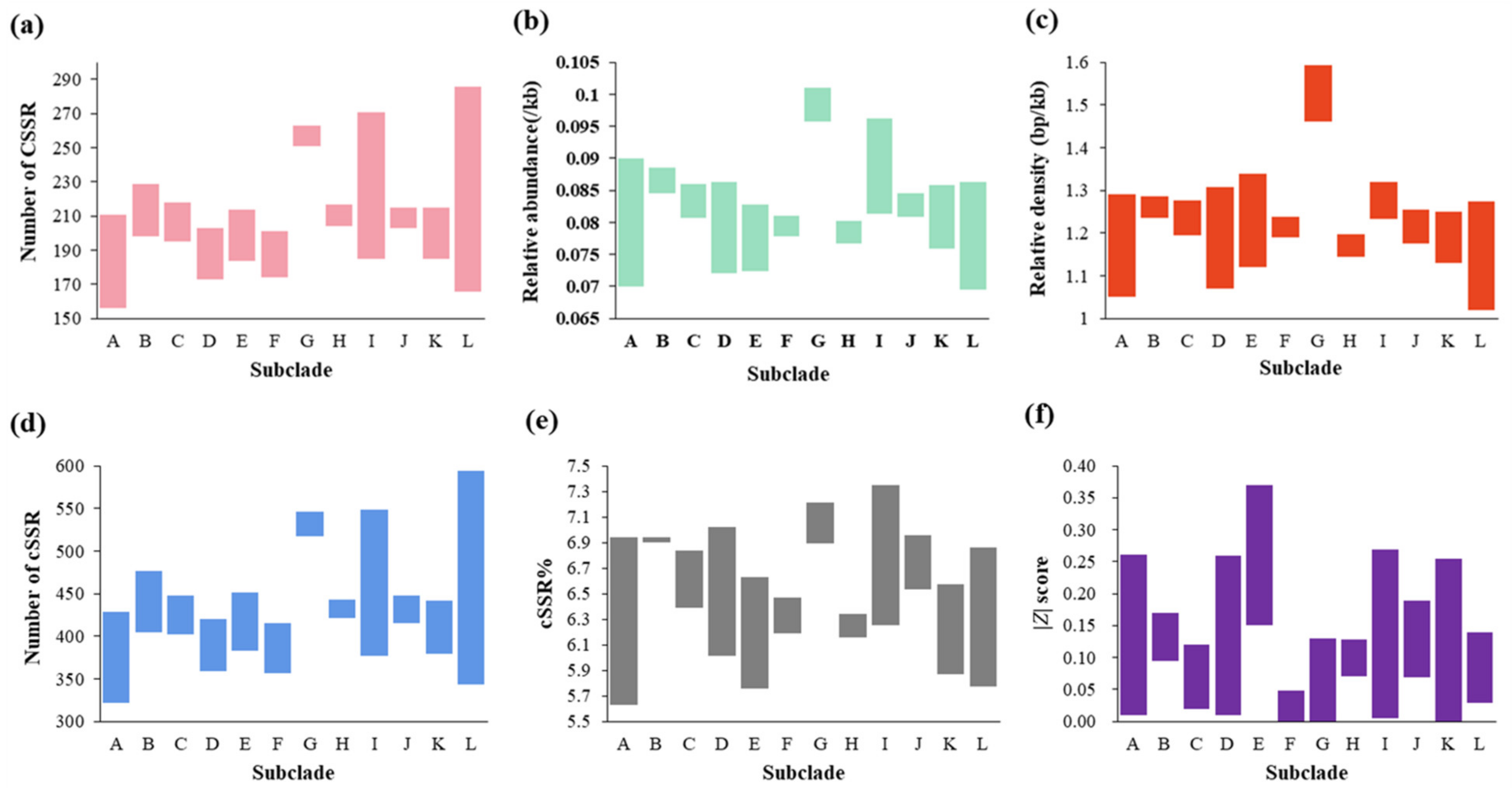
2.4. Complexity, Motifs, and Distribution of CSSRs
2.5. Pearson Linear Correlation
3. Discussion
4. Materials and Methods
4.1. Genome Sequences
4.2. Phylogenetic Analysis and Genome Comparison
4.3. Detection and Analysis of SSRs and CSSRs
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buitenhuis, E. Picophytoplankton biomass distribution in the global ocean. Earth Syst. Sci. Data 2012, 5, 221–242. [Google Scholar] [CrossRef] [Green Version]
- Dvořák, P.; Casamatta, D.A.; Poulíčková, A.; Hašler, P.; Ondřej, V.; Sanges, R. Synechococcus: 3 billion years of global dominance. Mol. Ecol. 2014, 23, 5538–5551. [Google Scholar] [CrossRef] [PubMed]
- Komárek, J.; Johansen, J.; Smarda, J.; Strunecký, O. Phylogeny and taxonomy of Synechococcus-like cyanobacteria. Fottea 2020, 20, 171–191. [Google Scholar] [CrossRef]
- Coutinho, F.; Tschoeke, D.A.; Thompson, F.; Thompson, C. Comparative genomics of Synechococcus and proposal of the new genus Parasynechococcus. PeerJ 2016, 4, e1522. [Google Scholar] [CrossRef] [Green Version]
- Rippka, R.; Cohen-Bazire, G. The cyanobacteriales: A legitimate order based on the type strain Cyanobacterium stanieri? Annales de l’Institut Pasteur/Microbiologie 1983, 134b, 21–36. [Google Scholar] [CrossRef]
- Shih, P.M.; Wu, D.; Latifi, A.; Axen, S.D.; Fewer, D.P.; Talla, E.; Calteau, A.; Cai, F.; Tandeau de Marsac, N.; Rippka, R.; et al. Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 1053–1058. [Google Scholar] [CrossRef] [Green Version]
- Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef]
- Oliveira, E.J.; Pádua, J.G.; Zucchi, M.I.; Vencovsky, R.; Vieira, M.L.C. Origin, evolution and genome distribution of microsatellites. Genet. Mol. Biol. 2006, 29, 294–307. [Google Scholar] [CrossRef]
- Yao, D.; Cheng, L.; Du, L.; Li, M.; Daroch, M.; Tang, J. Genome-wide investigation and analysis of microsatellites and compound microsatellites in Leptolyngbya-like species, Cyanobacteria. Life 2021, 11, 1258. [Google Scholar] [CrossRef]
- Kabra, R.; Kapil, A.; Attarwala, K.; Rai, P.K.; Shanker, A. Identification of common, unique and polymorphic microsatellites among 73 cyanobacterial genomes. World J. Microbiol. Biotechnol. 2016, 32, 71. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.F.; Wang, R.; Wang, S. Microsatellites in pursuit of microbial genome evolution. Front. Microbiol. 2016, 6, 1462. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.; Aertsen, A.; Michiels, C.W. The role of variable DNA tandem repeats in bacterial adaptation. FEMS Microbiol. Rev. 2014, 38, 119–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, H.; Zhang, P.; Shi, T.; Jian, Y.; Long, W.; Wang, S.; Su, Y.; Zhang, H.; Qiao, Y.; Li, X. Genome-wide characterization of simple sequence repeats in Pyrus bretschneideri and their application in an analysis of genetic diversity in pear. BMC Genomics 2018, 19, 473. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.P.; Jones, A.G. The future of parentage analysis: From microsatellites to SNPs and beyond. Mol. Ecol. 2019, 28, 544–567. [Google Scholar] [CrossRef] [PubMed]
- Ledenyova, M.L.; Tkachenko, G.A.; Shpak, I.M. Imperfect and compound microsatellites in the genomes of Burkholderia pseudomallei strains. Mol. Biol. 2019, 53, 127–137. [Google Scholar] [CrossRef]
- Evirgh, R.K.; Hedayat, N.; Hafezian, S.H.; Farhadi, A.; Bakhtiarizadeh, M.R. Genome-wide identification of microsatellites and transposable elements in the dromedary camel genome using whole-genome sequencing data. Front. Genet. 2019, 10, 692. [Google Scholar]
- Chen, M.-Y.; Teng, W.-K.; Zhao, L.; Hu, C.-X.; Zhou, Y.-K.; Han, B.-P.; Song, L.-R.; Shu, W.-S. Comparative genomics reveals insights into cyanobacterial evolution and habitat adaptation. ISME J. 2021, 15, 211–227. [Google Scholar] [CrossRef]
- Tang, J.; Li, L.; Li, M.; Du, L.; Shah, M.R.; Waleron, M.; Waleron, M.; Waleron, K.F.; Daroch, M. Description, taxonomy, and comparative genomics of a novel species, Thermoleptolyngbya sichuanensis sp. nov., isolated from Hot Springs of Ganzi, Sichuan, China. Front. Microbiol. 2021, 12, 696102. [Google Scholar] [CrossRef]
- Tang, J.; Du, L.; Li, M.; Yao, D.; Waleron, M.; Waleron, K.F.; Daroch, M. Characterization of a novel hot-spring cyanobacterium Leptodesmis sichuanensis sp. nov. and genomic insights of molecular adaptations into its habitat. Front. Microbiol. 2022, 12, 739625. [Google Scholar] [CrossRef]
- Komárek, J.; Kaštovský, J.; Mares, J.; Johansen, J. Taxonomic classification of cyanoprokaryotes (cyanobacterial genera) 2014, using a polyphasic approach. Preslia 2014, 86, 295–335. [Google Scholar]
- Herdman, M.; Castenholz, R.; Waterbury, J.; Rippka, R. Form-genus XIII. Synechococcus. In Bergey’s Manual of Systematic Bacteriology, 2nd ed.; Springer: New York, NY, USA, 2001; pp. 508–512. [Google Scholar]
- Jain, C.; Rodriguez, R.L. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [Green Version]
- Walter, J.M.; Coutinho, F.H.; Dutilh, B.E.; Swings, J.; Thompson, F.L.; Thompson, C.C. Ecogenomics and Taxonomy of Cyanobacteria Phylum. Front. Microbiol. 2017, 8, 2132. [Google Scholar] [CrossRef]
- Lapage, S.P.; Sneath, P.H.A.; Lessel, E.F.; Skerman, V.B.D.; Seeliger, H.P.R.; Clark, W.A. (Eds.) International Code of Nomenclature of Bacteria: Bacteriological Code, 1990 Revision; ASM Press: Washington, DC, USA, 1992. [Google Scholar]
- Turland, N.; Wiersema, J.; Barrie, F.R.; Greuter, W.; Smith, G.F. International Code of Nomenclature for Algae, Fungi, and Plants (Shenzhen Code) Adopted by the Nineteenth International Botanical Congress Shenzhen, China, July 2017; Koeltz Botanical Books: Glashütten, Germany, 2018. [Google Scholar]
- Tang, J.; Shah, M.R.; Yao, D.; Jiang, Y.; Du, L.; Zhao, K.; Li, L.; Li, M.; Waleron, M.; Waleron, M.; et al. Polyphasic identification and genomic insights of Leptothermofonsia sichuanensis gen. sp. nov., a novel thermophilic cyanobacteria within Leptolyngbyaceae. Front. Microbiol. 2022, 13, 765105. [Google Scholar] [CrossRef]
- Sciuto, K.; Moro, I. Detection of the new cosmopolitan genus Thermoleptolyngbya (Cyanobacteria, Leptolyngbyaceae) using the 16S rRNA gene and 16S–23S ITS region. Mol. Phylogenet. Evol. 2016, 105, 15–35. [Google Scholar] [CrossRef]
- Mrázek, J.; Guo, X.; Shah, A. Simple sequence repeats in prokaryotic genomes. Proc. Natl. Acad. Sci. USA 2007, 104, 8472–8477. [Google Scholar] [CrossRef] [Green Version]
- Mareš, J.; Strunecký, O.; Bučinská, L.; Wiedermannová, J. Evolutionary patterns of thylakoid architecture in cyanobacteria. Front. Microbiol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Dvořák, P. Genome-wide analysis of cyanobacterial evolution: The example of Synechococcus. In Cyanobacterial Omics and Manipulation; Los, D.A., Ed.; Caister Academic Press: Norfolk, UK, 2017; pp. 35–53. [Google Scholar]
- Dvořák, P.; Hindak, F.; Hašler, P.; Hindakova, A.; Poulíčková, A. Morphological and molecular studies of Neosynechococcus sphagnicola, gen. et sp. nov. (Cyanobacteria, Synechococcales). Phytotaxa 2014, 170, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Komárek, J.; Kopecký, J.; Cepák, V. Generic characters of the simplest cyanoprokaryotes Cyanobium, Cyanobacterium and Synechococcus. Cryptogam. Algol. 1999, 20, 209–222. [Google Scholar] [CrossRef]
- Kassai-Jáger, E.; Ortutay, C.; Tóth, G.; Vellai, T.; Gáspári, Z. Distribution and evolution of short tandem repeats in closely related bacterial genomes. Gene 2008, 410, 18–25. [Google Scholar] [CrossRef]
- Du, L.-M.; Liu, Q.; Zhao, K.-L.; Tang, J.; Fan, Z.-X. PSMD: An extensive database for pan-species microsatellite investigation and marker development. Mol. Ecol. Resour. 2020, 20, 283–291. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, M.; Wang, H.; Wang, J.F. Microsatellites in the genome of the edible mushroom, Volvariella volvacea. BioMed Res. Int. 2014, 2014, 281912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, S.; Mishra, R.K.; Singh, L. Genome-wide analysis of microsatellite repeats in humans: Their abundance and density in specific genomic regions. Genome Biol. 2003, 4, R13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Peng, M.; Fang, Z. The direction of microsatellite mutations is dependent upon allele length. Nat. Genet. 2000, 24, 396–399. [Google Scholar] [CrossRef]
- Li, Y.-C.; Korol, A.B.; Tzion, F.; Eviatar, N. Microsatellites within genes: Structure, function, and evolution. Mol. Biol. Evol. 2004, 21, 991–1007. [Google Scholar] [CrossRef]
- Trifonov, E.N. Tuning function of tandemly repeating sequences: A molecular device for fast adaptation. In Evolutionary Theory and Processes: Modern Horizons: Papers in Honour of Eviatar Nevo; Wasser, S.P., Ed.; Springer: Dordrecht, The Netherlands, 2004; pp. 115–138. [Google Scholar] [CrossRef]
- Bhargava, A.; Fuentes, F.F. Mutational dynamics of microsatellites. Mol. Biotechnol. 2010, 44, 250–266. [Google Scholar] [CrossRef]
- Li, Y.-C.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: Genomic distribution, putative functions and mutational mechanisms: A review. Mol. Ecol. 2002, 11, 2453–2465. [Google Scholar] [CrossRef]
- Hernández-López, A.; Chabrol, O.; Royer-Carenzi, M.; Merhej, V.; Pontarotti, P.; Raoult, D. To tree or not to tree? Genome-wide quantification of recombination and reticulate evolution during the diversification of strict intracellular bacteria. Genome Biol. Evol. 2013, 5, 2305–2317. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Du, L.; Liang, Y.; Daroch, M. Complete genome sequence and comparative analysis of Synechococcus sp. CS-601 (SynAce01), a cold-adapted cyanobacterium from an oligotrophic Antarctic habitat. Int. J. Mol. Sci. 2019, 20, 152. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Jiang, D.; Luo, Y.; Liang, Y.; Li, L.; Shah, M.M.R.; Daroch, M. Potential new genera of cyanobacterial strains isolated from thermal springs of western Sichuan, China. Algal Res. 2018, 31, 14–20. [Google Scholar] [CrossRef]
- Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Chi, Z.; Qin, L.; Zhang, X.; Yue, B. Krait: An ultrafast tool for genome-wide survey of microsatellites and primer design. Bioinformatics 2017, 34, 681–683. [Google Scholar] [CrossRef]
- Jan, M.z. Analysis of distribution indicates diverse functions of simple sequence repeats in Mycoplasma Genomes. Mol. Biol. Evol. 2006, 23, 1370–1385. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subclade | Strain | M16.1 | 8L107 | PROS-U-1 | WH 8102 | KORDI-49 | BS56D | WH 8101 | UW105 | MVIR-18-1 | A18-25c | WH 7803 | BIOS-E4-1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | M16.1 | 100 | |||||||||||
| B | 8L107 | 77.5 | 100 | ||||||||||
| C | PROS-U-1 | 82.0 | 77.5 | 100 | |||||||||
| D | WH 8102 | 80.6 | 77.7 | 80.2 | 100 | ||||||||
| E | KORDI-49 | 79.9 | 78.2 | 79.6 | 80.1 | 100 | |||||||
| F | BS56D | 78.9 | 78.0 | 79.8 | 79.0 | 78.8 | 100 | ||||||
| G | WH 8101 | 78.6 | 77.7 | 78.6 | 78.9 | 78.6 | 81.7 | 100 | |||||
| H | UW105 | 78.5 | 77.9 | 78.7 | 78.1 | 78.0 | 78.2 | 77.8 | 100 | ||||
| I | MVIR-18-1 | 77.9 | 78.9 | 78.2 | 78.5 | 77.7 | 77.8 | 77.8 | 77.5 | 100 | |||
| J | A18-25c | 80.3 | 77.9 | 80.0 | 79.7 | 79.0 | 78.9 | 78.6 | 78.3 | 77.6 | 100 | ||
| K | WH 7803 | 79.7 | 77.9 | 79.7 | 80.3 | 79.6 | 79.1 | 79.0 | 78.0 | 78.0 | 79.3 | 100 | |
| L | BIOS-E4-1 | 78.8 | 78.2 | 78.8 | 78.3 | 78.9 | 78.8 | 78.0 | 78.3 | 78.1 | 78.2 | 78.0 | 100 |
| Genome Size | GC Content | nSSR | ncSSR | ||
|---|---|---|---|---|---|
| nSSR | ρ | 0.96 | −0.40 | - | 0.91 |
| Significance | p < 0.01 | p < 0.01 | - | p < 0.01 | |
| nCSSR | ρ | 0.82 | −0.40 | 0.91 | 1.00 |
| Significance | p < 0.01 | p < 0.01 | p < 0.01 | p < 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, J.; Yao, D.; Zhou, H.; Du, L.; Daroch, M. Reevaluation of Parasynechococcus-like Strains and Genomic Analysis of Their Microsatellites and Compound Microsatellites. Plants 2022, 11, 1060. https://doi.org/10.3390/plants11081060
Tang J, Yao D, Zhou H, Du L, Daroch M. Reevaluation of Parasynechococcus-like Strains and Genomic Analysis of Their Microsatellites and Compound Microsatellites. Plants. 2022; 11(8):1060. https://doi.org/10.3390/plants11081060
Chicago/Turabian StyleTang, Jie, Dan Yao, Huizhen Zhou, Lianming Du, and Maurycy Daroch. 2022. "Reevaluation of Parasynechococcus-like Strains and Genomic Analysis of Their Microsatellites and Compound Microsatellites" Plants 11, no. 8: 1060. https://doi.org/10.3390/plants11081060
APA StyleTang, J., Yao, D., Zhou, H., Du, L., & Daroch, M. (2022). Reevaluation of Parasynechococcus-like Strains and Genomic Analysis of Their Microsatellites and Compound Microsatellites. Plants, 11(8), 1060. https://doi.org/10.3390/plants11081060