
An Integration of MicroRNA and Transcriptome Sequencing Analysis Reveal Regulatory Roles of miRNAs in Response to Chilling Stress in Wild Rice


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification of miRNAs That Are Involved in Chilling Stress Response via Small RNA Sequencing
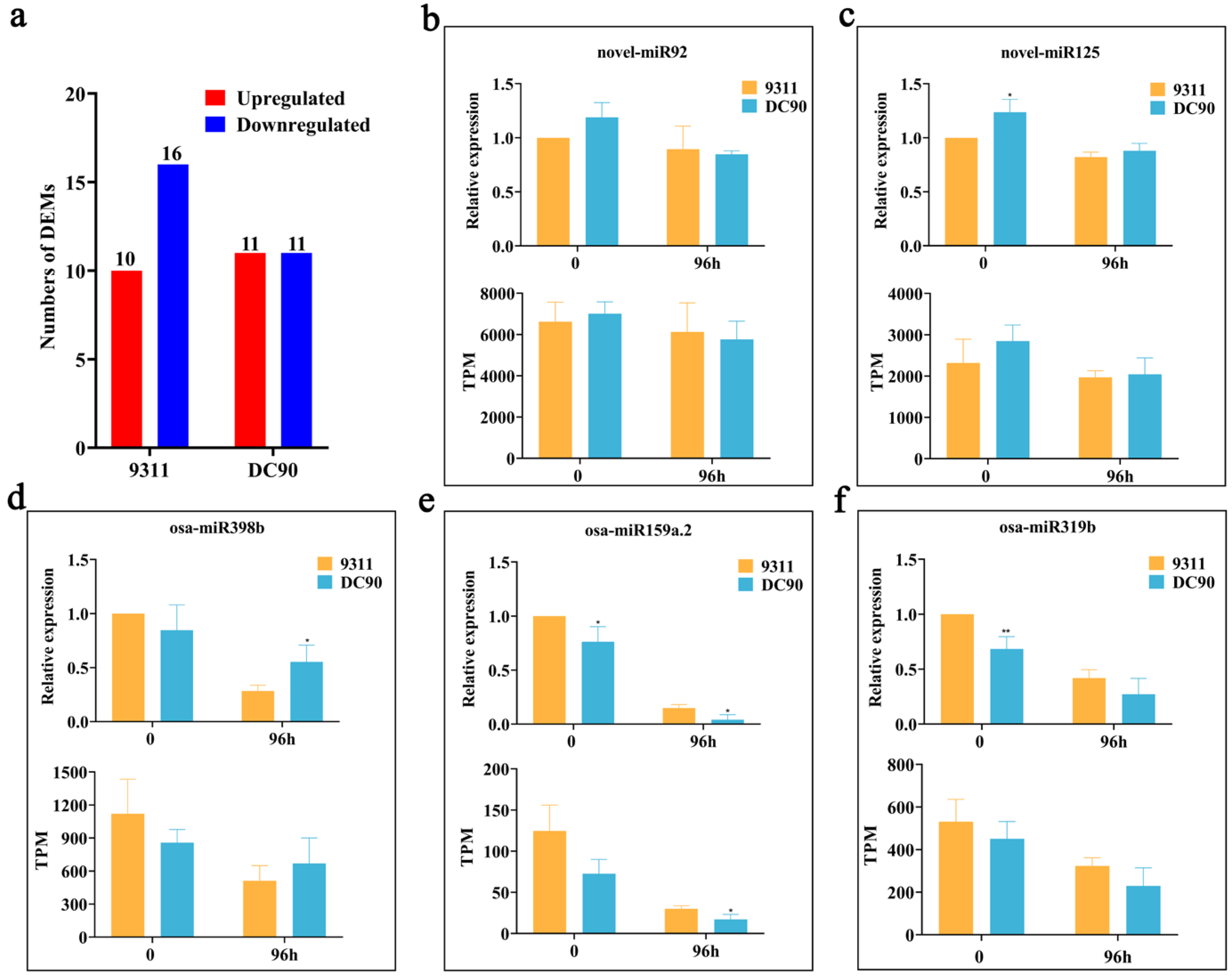
2.2. Expression Profile Analysis of Chilling-Induced miRNAs
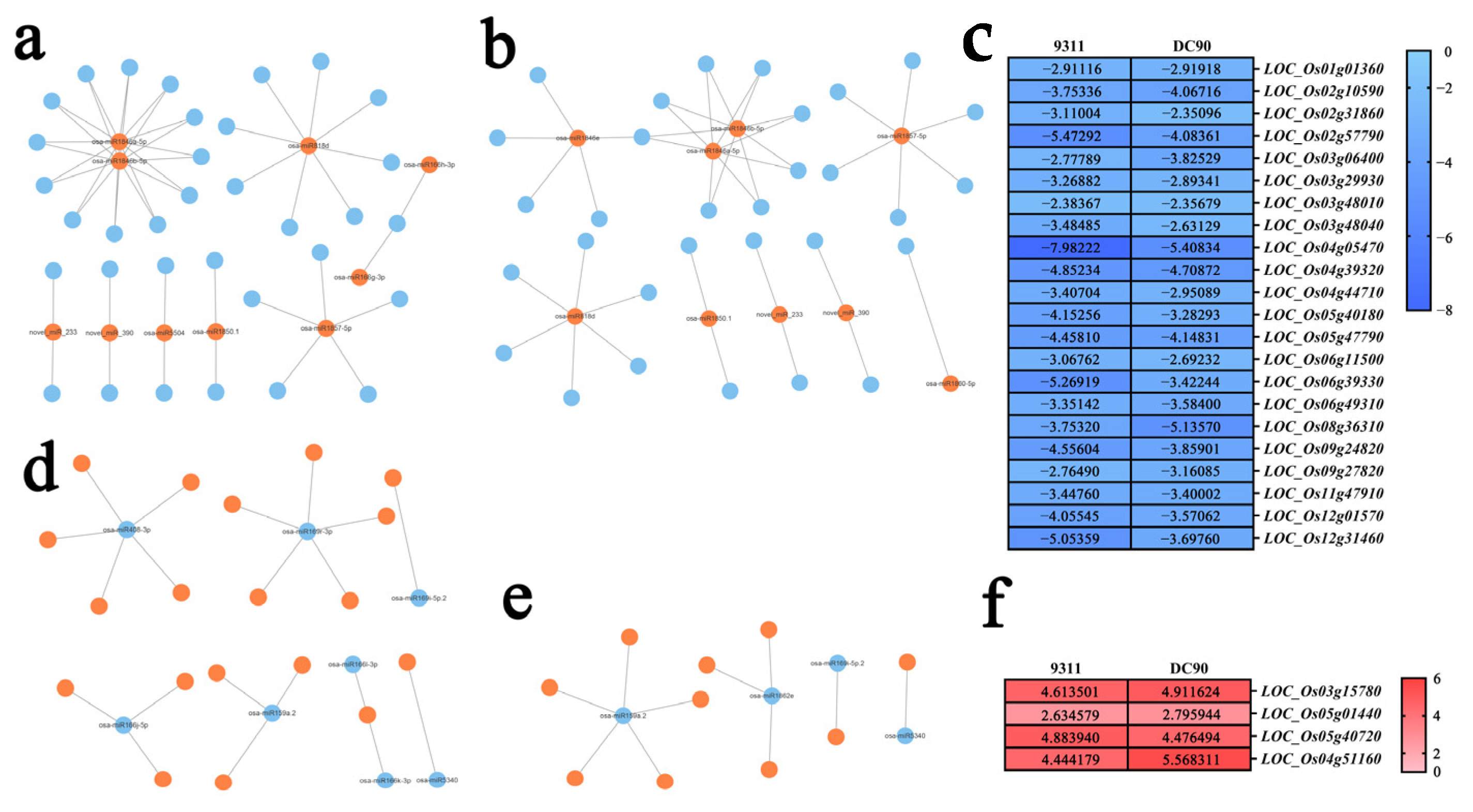
2.3. Integration of mRNA-Seq and miRNA-Seq to Predict Target Genes of the DEMs
2.4. Analysis of the Regulatory Relationship between DEMs and Target Genes Based on the Expression Regulatory Networks
2.5. KEGG Enrichment Analysis of DEM Target Genes
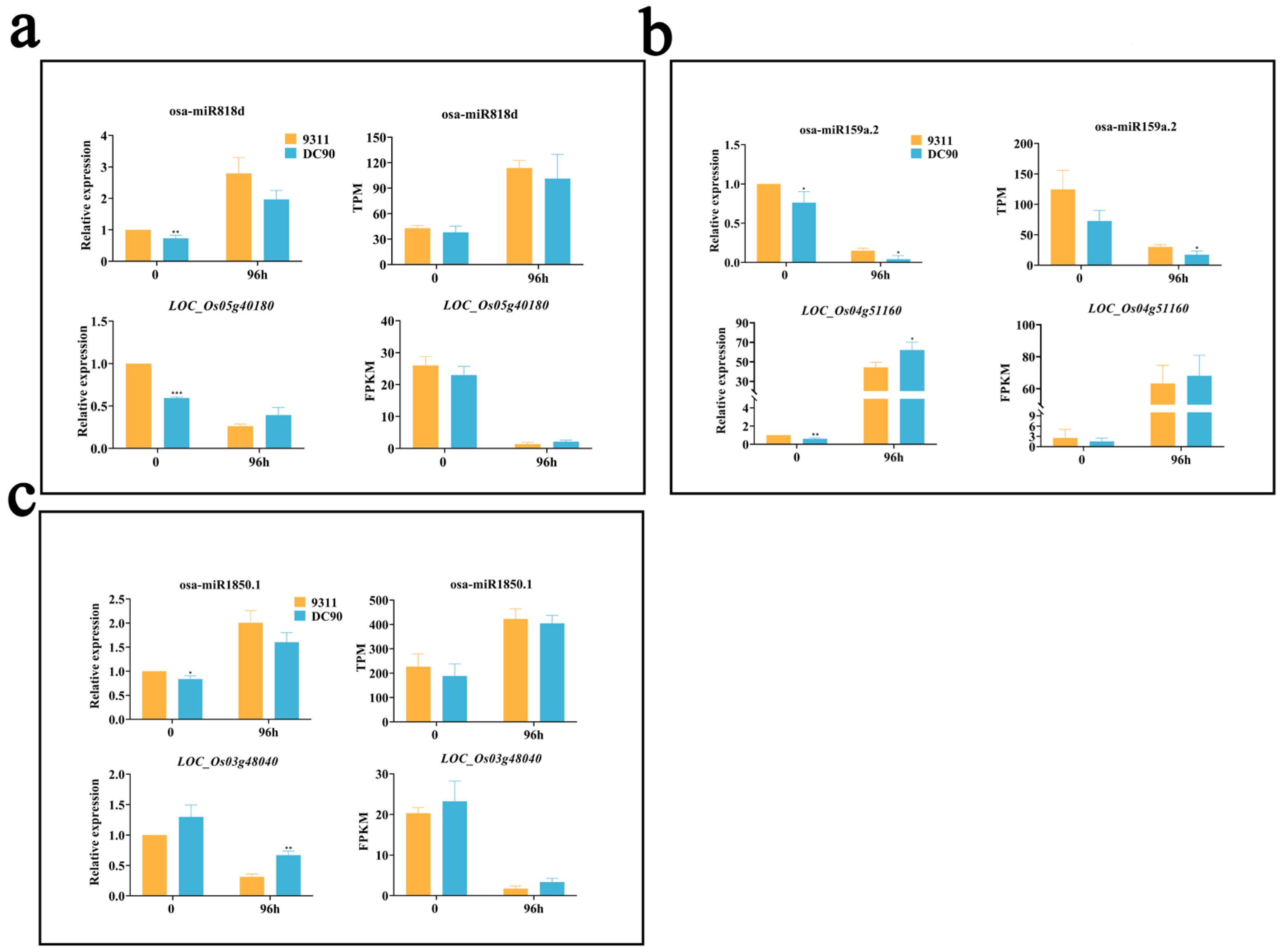
2.6. Validation of the Regulatory Relationship between DEMs and SDETGs
2.7. CRISPR/Cas9-Edited Putative SDETG, OsGL1-11, Leads to Chilling Sensitive Phenotype in Rice
3. Discussion
4. Materials and Methods
4.1. Plant Materials, Growth Conditions, and Chilling Stress Treatment
4.2. High-Throughput Deep Sequencing of Small RNA
4.3. Transcriptomic Profiling
4.4. Bioinformatics Analysis of Small RNA Sequencing Data
4.5. Quantification of miRNA Expression Level by qRT-PCR
4.6. Generation of CRISPR/Cas9 Knockout Lines
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, J.K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Pareek, A.; Khurana, A.; Sharma, A.K.; Kumar, R. An overview of signaling regulons during cold stress tolerance in plants. Curr. Genom. 2017, 18, 498–511. [Google Scholar] [CrossRef]
- Solanke, A.U.; Sharma, A.K. Signal transduction during cold stress in plants. Physiol. Mol. Biol. Plants 2008, 14, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, S.K.; Pandit, E.; Nayak, D.K.; Behera, L.; Mohapatra, T. Genes, pathways and transcription factors involved in seedling stage chilling stress tolerance in indica rice through RNA-Seq analysis. BMC Plant Biol. 2019, 19, 352. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Yu, S.; Kong, D.; Xiong, J.; Ma, X.; Chen, L.; Luo, L. Temporal responses of conserved miRNAs to drought and their associations with drought tolerance and productivity in rice. BMC Genom. 2020, 21, 232. [Google Scholar] [CrossRef] [Green Version]
- Pandit, E.; Tasleem, S.; Barik, S.R.; Mohanty, D.P.; Nayak, D.K.; Mohanty, S.P.; Das, S.; Pradhan, S.K. Genome-wide association mapping reveals multiple QTLs governing tolerance response for seedling stage chilling stress in Indica Rice. Front Plant Sci. 2017, 8, 552. [Google Scholar] [CrossRef] [Green Version]
- Kayess, M.O.; Hassan, M.M.; Nurhasan, M.; Ahmed, K. Effect of low temperature on chlorophyll and carotenoid content on the seedlings of some selected boro rice varieties. Am. J. Plant Sci. 2020, 11, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Fujino, K.; Sekiguchi, H.; Sato, T.; Kiuchi, H.; Nonoue, Y.; Takeuchi, Y.; Ando, T.; Lin, S.Y.; Yano, M. Mapping of quantitative trait loci controlling low-temperature germinability in rice (Oryza sativa L.). Theor. Appl. Genet. 2004, 108, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Andaya, V.C.; Mackill, D.J. Mapping of QTLs associated with cold tolerance during the vegetative stage in rice. J. Exp. Bot. 2003, 54, 2579–2585. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.P.; Jeung, J.U.; Lee, J.I.; Choi, Y.H.; Yea, J.D.; Virk, P.S.; Mackill, D.J.; Jena, K.K. Identification and analysis of QTLs controlling cold tolerance at the reproductive stage and validation of effective QTLs in cold-tolerant genotypes of rice (Oryza sativa L.). Theor. Appl. Genet. 2010, 120, 985–995. [Google Scholar] [CrossRef]
- Andaya, V.C.; Tai, T.H. Fine mapping of the qCTS12 locus, a major QTL for seedling cold tolerance in rice. Theor. Appl. Genet. 2006, 113, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Das, A.; Datta, S.; Banerjee, I.; Tripathy, S.; Chaudhuri, S. Understanding the early cold response mechanism in IR64 indica rice variety through comparative transcriptome analysis. BMC Genom. 2020, 21, 425. [Google Scholar] [CrossRef]
- Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [Green Version]
- Sun, G. MicroRNAs and their diverse functions in plants. Plant Mol. Biol. 2011, 80, 17–36. [Google Scholar] [CrossRef]
- Gielen, H.; Remans, T.; Vangronsveld, J.; Cuypers, A. MicroRNAs in metal stress: Specific roles or secondary responses? Int. J. Mol. Sci. 2012, 13, 15826–15847. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Mohapatra, R.; Mahanty, B.; Joshi, R.K. Analysis of microRNAs and their targets from onion (Allium cepa) using genome survey sequences (GSS) and expressed sequence tags (ESTs). Bioinformation 2019, 15, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Leung, A.K.; Sharp, P.A. MicroRNA functions in stress responses. Mol. Cell 2010, 40, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhang, B. MicroRNAs in control of plant development. J. Cell. Physiol. 2016, 231, 303–313. [Google Scholar] [CrossRef]
- Khraiwesh, B.; Zhu, J.K.; Zhu, J. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim. Biophys. Acta 2012, 1819, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Xie, K.; Xiong, L. Conserved miR164-targeted NAC genes negatively regulate drought resistance in rice. J. Exp. Bot. 2014, 65, 2119–2135. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Li, D.; Mao, D.; Liu, X.U.E.; Ji, C.; Li, X.; Zhao, X.; Cheng, Z.; Chen, C.; Zhu, L. Overexpression of microRNA319 impacts leaf morphogenesis and leads to enhanced cold tolerance in rice (Oryza sativa L.). Plant Cell Environ. 2013, 36, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Unver, T.; Wang, S.-t.; Sun, X.-l.; Hoshino, Y.; Yu, Y.; Jia, B.; Sun, Z.-w.; Sun, M.-z.; Duan, X.-b.; Zhu, Y.-m. MicroRNA319 positively regulates cold tolerance by targeting OsPCF6 and OsTCP21 in rice (Oryza sativa L.). PLoS ONE 2014, 9, e91357. [Google Scholar]
- Zhou, M.; Tang, W. MicroRNA156 amplifies transcription factor-associated cold stress tolerance in plant cells. Mol. Genet. Genom. 2019, 294, 379–393. [Google Scholar] [CrossRef]
- Cui, N.; Sun, X.; Sun, M.; Jia, B.; Duanmu, H.; Lv, D.; Duan, X.; Zhu, Y. Overexpression of OsmiR156k leads to reduced tolerance to cold stress in rice (Oryza Sativa). Mol. Breed. 2015, 35, 214. [Google Scholar] [CrossRef]
- Sun, M.; Yang, J.; Cai, X.; Shen, Y.; Cui, N.; Zhu, Y.; Jia, B.; Sun, X. The opposite roles of OsmiR408 in cold and drought stress responses in Oryza sativa. Mol. Breed. 2018, 38, 120. [Google Scholar] [CrossRef]
- Sun, M.; Shen, Y.; Yang, J.; Cai, X.; Li, H.; Zhu, Y.; Jia, B.; Sun, X. miR535 negatively regulates cold tolerance in rice. Mol. Breed. 2020, 40, 14. [Google Scholar] [CrossRef]
- Sunkar, R.; Zhou, X.; Zheng, Y.; Zhang, W.; Zhu, J.K. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 2008, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Shi, W.; Ma, X.; Zhao, J.; Wang, S.; Tan, L.; Sun, C.; Liu, F. Identification of microRNAs responding to cold stress in Dongxiang common wild rice. Genome 2019, 62, 635–642. [Google Scholar] [CrossRef]
- Song, G.; Zhang, R.; Zhang, S.; Li, Y.; Gao, J.; Han, X.; Chen, M.; Wang, J.; Li, W.; Li, G. Response of microRNAs to cold treatment in the young spikes of common wheat. BMC Genom. 2017, 18, 212. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xu, Y.; Huan, Q.; Chong, K. Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genom. 2009, 10, 449. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Sun, Y.H.; Chiang, V.L. Stress-responsive microRNAs in Populus. Plant J. 2008, 55, 131–151. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.H.; Tian, X.; Li, Y.J.; Wu, C.A.; Zheng, C.C. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 2008, 14, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cen, W.; Zhao, W.; Ma, M.; Lu, S.; Liu, J.; Cao, Y.; Zeng, Z.; Wei, H.; Wang, S.; Li, R.; et al. The wild rice locus CTS-12 mediates ABA-dependent stomatal opening modulation to limit water loss under severe chilling stress. Front. Plant Sci. 2020, 11, 575699. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome. Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- He, L.; Li, M.; Qiu, Z.; Chen, D.; Zhang, G.; Wang, X.; Chen, G.; Hu, J.; Gao, Z.; Dong, G.; et al. Primary leaf-type ferredoxin 1 participates in photosynthetic electron transport and carbon assimilation in rice. Plant J. 2020, 104, 44–58. [Google Scholar] [CrossRef]
- Li, C.; Hu, Y.; Huang, R.; Ma, X.; Wang, Y.; Liao, T.; Zhong, P.; Xiao, F.; Sun, C.; Xu, Z.; et al. Mutation of FdC2 gene encoding a ferredoxin-like protein with C-terminal extension causes yellow-green leaf phenotype in rice. Plant Sci. 2015, 238, 127–134. [Google Scholar] [CrossRef]
- Nath, K.; Poudyal, R.S.; Eom, J.S.; Park, Y.S.; Zulfugarov, I.S.; Mishra, S.R.; Tovuu, A.; Ryoo, N.; Yoon, H.S.; Nam, H.G.; et al. Loss-of-function of OsSTN8 suppresses the photosystem II core protein phosphorylation and interferes with the photosystem II repair mechanism in rice (Oryza sativa). Plant J. 2013, 76, 675–686. [Google Scholar] [CrossRef]
- Islam, M.A.; Du, H.; Ning, J.; Ye, H.; Xiong, L. Characterization of Glossy1-homologous genes in rice involved in leaf wax accumulation and drought resistance. Plant Mol. Biol. 2009, 70, 443–456. [Google Scholar] [CrossRef]
- Zhou, L.; Ni, E.; Yang, J.; Zhou, H.; Liang, H.; Li, J.; Jiang, D.; Wang, Z.; Liu, Z.; Zhuang, C. Rice OsGL1-6 is involved in leaf cuticular wax accumulation and drought resistance. PLoS ONE 2013, 8, e65139. [Google Scholar] [CrossRef]
- Kurokawa, Y.; Nagai, K.; Huan, P.D.; Shimazaki, K.; Qu, H.; Mori, Y.; Toda, Y.; Kuroha, T.; Hayashi, N.; Aiga, S.; et al. Rice leaf hydrophobicity and gas films are conferred by a wax synthesis gene (LGF1) and contribute to flood tolerance. New Phytol. 2018, 218, 1558–1569. [Google Scholar] [CrossRef] [Green Version]
- Qin, B.X.; Tang, D.; Huang, J.; Li, M.; Wu, X.R.; Lu, L.L.; Wang, K.J.; Yu, H.X.; Chen, J.M.; Gu, M.H.; et al. Rice OsGL1-1 is involved in leaf cuticular wax and cuticle membrane. Mol. Plant 2011, 4, 985–995. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Burd, S.; Lers, A. miR408 is involved in abiotic stress responses in Arabidopsis. Plant J. 2015, 84, 169–187. [Google Scholar] [CrossRef]
- Pan, J.; Huang, D.; Guo, Z.; Kuang, Z.; Zhang, H.; Xie, X.; Ma, Z.; Gao, S.; Lerdau, M.T.; Chu, C.; et al. Overexpression of microRNA408 enhances photosynthesis, growth, and seed yield in diverse plants. J. Integr. Plant Biol. 2018, 60, 323–340. [Google Scholar] [CrossRef]
- Axtell, M.J.; Bowman, J.L. Evolution of plant microRNAs and their targets. Trends Plant Sci. 2008, 13, 343–349. [Google Scholar] [CrossRef]
- Salvador-Guirao, R.; Hsing, Y.I.; San Segundo, B. The polycistronic miR166k-166h positively regulates rice immunity via post-transcriptional control of EIN2. Front. Plant Sci. 2018, 9, 337. [Google Scholar] [CrossRef]
- Ding, Y.; Gong, S.; Wang, Y.; Wang, F.; Bao, H.; Sun, J.; Cai, C.; Yi, K.; Chen, Z.; Zhu, C. MicroRNA166 modulates cadmium tolerance and accumulation in Rice. Plant Physiol. 2018, 177, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, H.; Srivastava, A.K.; Pan, Y.; Bai, J.; Fang, J.; Shi, H.; Zhu, J.K. Knockdown of rice MicroRNA166 confers drought resistance by causing leaf rolling and altering stem xylem development. Plant Physiol. 2018, 176, 2082–2094. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Ge, L.; Liang, R.; Li, W.; Ruan, K.; Lin, H.; Jin, Y. Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Mol. Biol. 2009, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Zeng, J.; He, X.-Q. MiR169 and its target PagHAP2-6 regulated by ABA are involved in poplar cambium dormancy. J. Plant Physiol. 2016, 198, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Balyan, S.; Jha, S.; Mathur, S. Novel insights into expansion and functional diversification of MIR169 family in tomato. Planta 2020, 251, 55. [Google Scholar] [CrossRef]
- Kan, Y.; Mu, X.-R.; Zhang, H.; Gao, J.; Shan, J.-X.; Ye, W.-W.; Lin, H.-X. TT2 controls rice thermotolerance through SCT1-dependent alteration of wax biosynthesis. Nat. Plants 2022, 8, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Cen, W.; Liu, J.; Lu, S.; Jia, P.; Yu, K.; Han, Y.; Li, R.; Luo, J. Comparative proteomic analysis of QTL CTS-12 derived from wild rice (Oryza rufipogon Griff.), in the regulation of cold acclimation and de-acclimation of rice (Oryza sativa L.) in response to severe chilling stress. BMC Plant Biol. 2018, 18, 163. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Jiang, L.; Wang, J.; Gu, P.; Chen, M. MTide: An integrated tool for the identification of miRNA-target interaction in plants. Bioinformatics 2015, 31, 290–291. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, Q.; Zhu, Q.; Liu, W.; Chen, Y.; Qiu, R.; Wang, B.; Yang, Z.; Li, H.; Lin, Y.; et al. A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot Plants. Mol. Plant 2015, 8, 1274–1284. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, W.; Xiao, W.; Sun, J.; Chen, M.; Ma, M.; Cao, Y.; Cen, W.; Li, R.; Luo, J. An Integration of MicroRNA and Transcriptome Sequencing Analysis Reveal Regulatory Roles of miRNAs in Response to Chilling Stress in Wild Rice. Plants 2022, 11, 977. https://doi.org/10.3390/plants11070977
Zhao W, Xiao W, Sun J, Chen M, Ma M, Cao Y, Cen W, Li R, Luo J. An Integration of MicroRNA and Transcriptome Sequencing Analysis Reveal Regulatory Roles of miRNAs in Response to Chilling Stress in Wild Rice. Plants. 2022; 11(7):977. https://doi.org/10.3390/plants11070977
Chicago/Turabian StyleZhao, Wenlong, Weiyu Xiao, Jinliang Sun, Mingxin Chen, Mingqing Ma, Yaqi Cao, Weijian Cen, Rongbai Li, and Jijing Luo. 2022. "An Integration of MicroRNA and Transcriptome Sequencing Analysis Reveal Regulatory Roles of miRNAs in Response to Chilling Stress in Wild Rice" Plants 11, no. 7: 977. https://doi.org/10.3390/plants11070977
APA StyleZhao, W., Xiao, W., Sun, J., Chen, M., Ma, M., Cao, Y., Cen, W., Li, R., & Luo, J. (2022). An Integration of MicroRNA and Transcriptome Sequencing Analysis Reveal Regulatory Roles of miRNAs in Response to Chilling Stress in Wild Rice. Plants, 11(7), 977. https://doi.org/10.3390/plants11070977