
The Grapevine Microbiome to the Rescue: Implications for the Biocontrol of Trunk Diseases

 , , ,
, , ,
Abstract
:1. Introduction
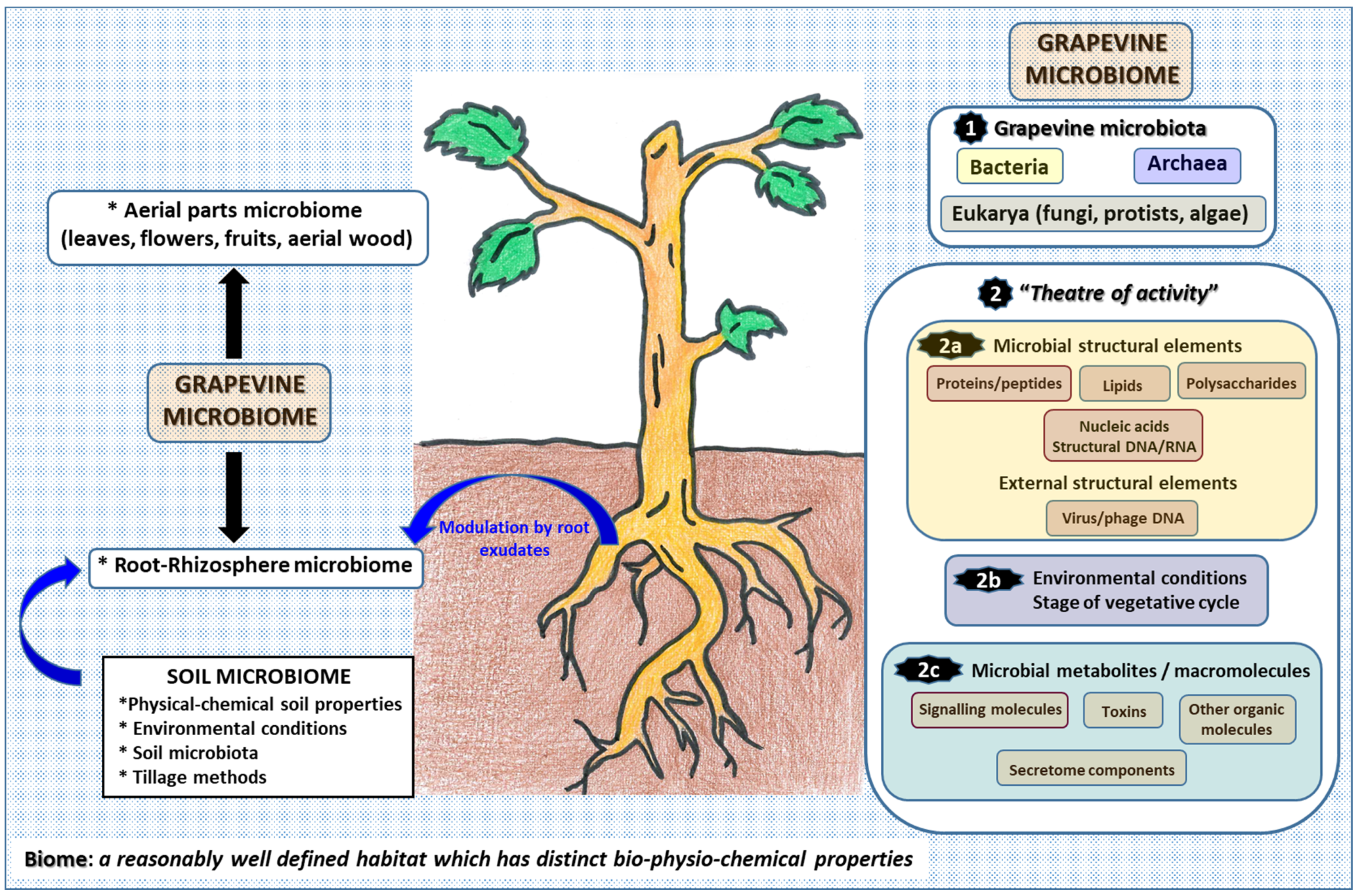
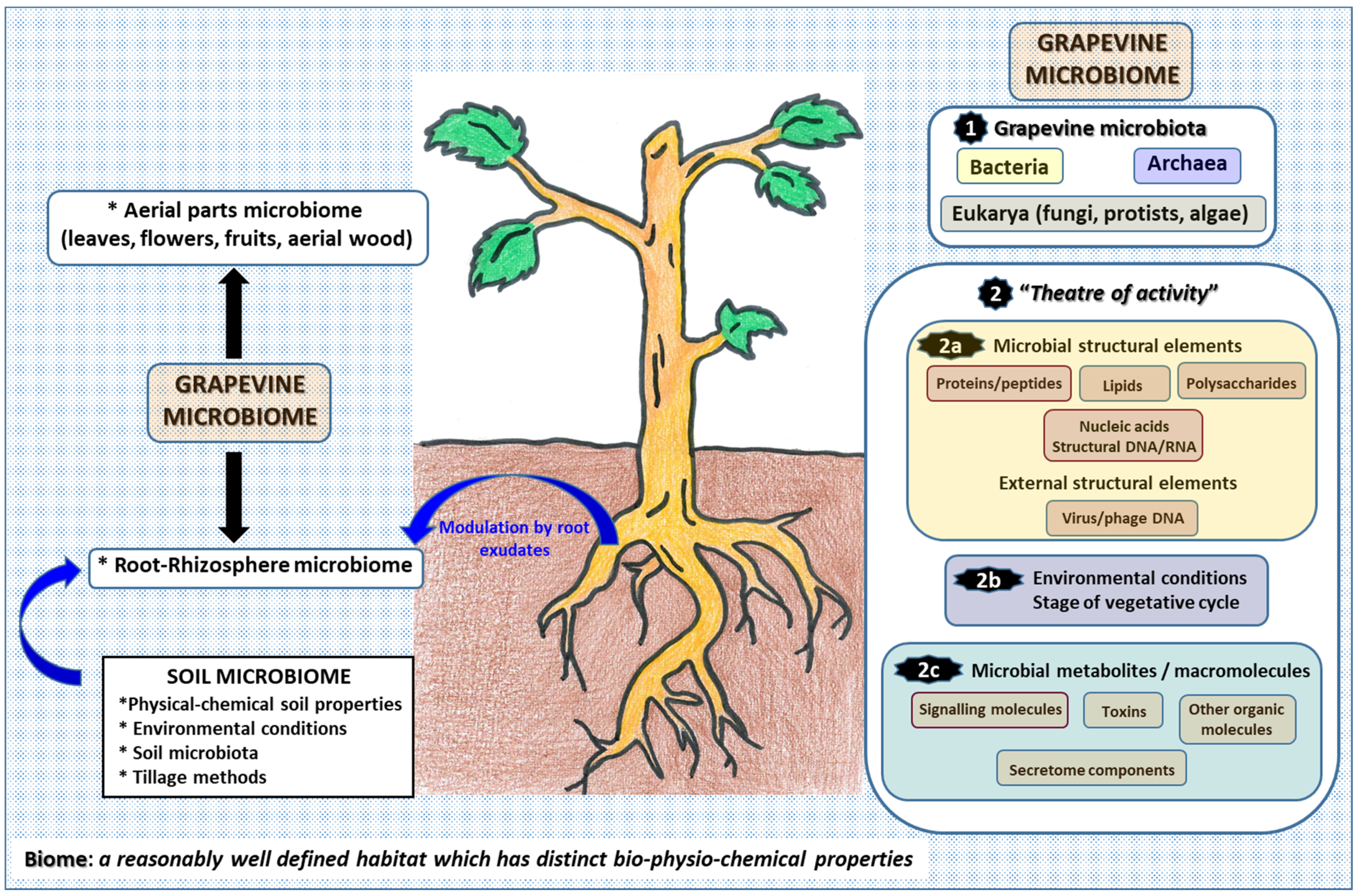
2. Insights into the Grapevine Microbiome: The Microbiome Concept
3. The Microbiome of Aerial Parts (Leaves, Flowers and Berries)

{kind=link}
{kind=link}
| Tissue/Material Analyzed | Grapevine/Rootstock Variety * | Geographical Area | Analysis Type | Microbiome Analyzed # | Year/Reference |
|---|---|---|---|---|---|
| Leaves | Tempranillo | Cantanhede, Portugal | Pyrosequencing | B, F | 2014 [12] |
| Leaves, flowers, grapes, roots, rhizosphere, bulk soil | Merlot | Long Island, NY, USA | Illumina NGS | B | 2015 [13] |
| Grapes | Carignan, Grenache | Priorat region, Tarragona, Spain | Illumina NGS | B | 2016 [15] |
| Grapes | Corvina | Gargagnago di Sant’Ambrogio di Valpolicella, Italy | Illumina NGS | B, F | 2016 [16] |
| Bulk soil | ND | Napa Valley, CA, USA | Illumina NGS | B | 2016 [17] |
| Bulk soil | ND | Central Chile | Roche 454 Gs Junior Titanium Series | B, F | 2017 [18] |
| Bulk soil | Cabernet Franc | Trentino South Tyrol (Italy) | Roche Gs FLX+ system | B, F | 2017 [19] |
| Bulk soil, rhizosphere | Pinot Noir | Carpeneto, Italy | Roche 454 Pyros. | B | 2017 [20] |
| Bulk soil, rhizosphere | Zweigelt grafted on BB5 | Lake Neusiedl, Austria | Illumina NGS | B | 2017 [21] |
| Grapes, bulk soil | Riesling | Ovid, NY, USA | Illumina NGS | B, F | 2018 [22] |
| Bulk soil, rhizosphere, root endosphere | Lambrusco grafted on 5BB, 1103P | Finale Emilia, Modena, Italy | Illumina NGS | B | 2018 [23] |
| Bulk soil | Riesling | Geinsenheim, Germany | Illumina NGS | B, F | 2018 [24] |
| Shoots, leaves, flowers, bark, root | Red Globe, Cabernet Gernischt | Beijing and Yunnan (China) | Illumina NGS | F | 2018 [25] |
| Wood | Midnight beauty | Beijing (China) | Illumina NGS | F | 2018 [26] |
| Wood | Pinot Meunier | Sonoma county (CA, USA) | Metatranscriptomic | F | 2018 [27] |
| Wood | Sauvignon Blanc, Grenache | Czech Republic, Spain | Metatranscriptomic | F | 2018 [28] |
| Rhizosphere, roots | Barbera grafted on 402A, 157.11, SO4, 161.49C | Oltrepo Pavese (Italy) | Illumina NGS | B | 2018 [29] |
| Wood | Cabernet Sauvignon | Lisbon (Portugal) | Illumina NGS | F | 2019 [30] |
| Rhizosphere | 110R, 140Ru, 1103P, 41B, 161-49C | Aldeanueva de Ebro and Olite (Spain) | Illumina NGS | B, F | 2019 [31] |
| Bulk soil, rhizosphere, endorhizosphere | Tempranillo | Las Rioja (Spain) | Illumina NGS | F | 2019 [32] |
| Wood | Verdelho, Shiraz | Hilltops and Hunter Valley. New South Wales (Australia) | Illumina NGS | B, F | 2020 [33] |
| Rhizosphere | 1103P, 140 Ru, 161-49 C, Kober 5BB | Rheingau, Germany | Ion Torrent Seq. | B | 2021 [34] |
| Bulk soil, rhizosphere, root, cordon, cane, sap | Syrah | Temecula (CA, USA) | Illumina NGS | B, F | 2020 [35] |
| Wood | Xinomavro, Agiorgitiko, Vidiano | NW (Amyntaio), central-south (Nemea) and southern Greece (Crete) | Illumina NGS | B, F | 2021 [36] |
| Wood | Malbec | Luján de Cuyo (Argentina) | Metatranscriptomic | B, F | 2021 [37] |
| Wood | 16 varieties grafted on 4 rootstocks | Cataluña (Spain) | Illumina NGS | F | 2022 [38] |
4. The Endomicrobiome and Wood Microbiome
4.1. The Microbiome of Grapevine Wood Endophytic Bacteria
4.2. The Endophytic Mycobiome of Grapevine Wood of Aerial Parts
4.3. The Endophytic Mycobiome of Grapevine Wood According to Metatranscriptomic Approaches
5. The Microbiome of Soil, Rhizosphere and Root Compartments
5.1. The Microbiome of Vineyard Soils and Its Relationship to the Rhizosphere Microbiome
5.2. The Microbiome of Grapevine Rootstocks
6. The Grapevine Microbiome as a Source of More Efficient and Promising BCAs against GTDs
6.1. Actinobacteria as Promising BCAs against Soil-Borne Fungal Pathogens Causing GTDs
| BCA | Source | Efficacy against GTD Pathogens in in Plant Trials | Reference |
|---|---|---|---|
| Bacillus pumilus (S32), Paenibacillus sp. (S19) | Wood tissue | Reduction of necrosis length caused by P. chlamydospora and induction of systemic resistance in planta (B. pumilus) | [78] |
| Pythium oligandrum | Rhizosphere | Significant reduction (40–50%) of necrosis in cv. Cabernet Sauvignon cuttings caused by P. chlamydospora in greenhouse assays | [79] |
| Streptomyces sp. VV/E1, VV/E2 and S. peucetius VV/E5 | Root endophytes | Significant decrease in infection level of grapevine grafts in nursery by Dactylonectria sp., Ilyonectria sp., P. chlamydospora and Ph. minimum | [77] |
| Streptomyces sp. VV/R1, VV/R4 and VV/R5 | Rhizosphere soil | Significant decrease in infection level of grapevine grafts in nursery by Dactylonectria sp., Ilyonectria sp., P. chlamydospora and Ph. minimum | [77] |
| Pythium oligandrum | Rhizosphere | Important reduction of necrosis lengths within the scion stem caused by N. parvum and P. chlamydospora | [80] |
| Pantoea agglomerans (S1); Paenibacillus sp. (S19) | Wood tissues (S19) or grape berries (S1) | Significant reduction of internal necrosis caused by N. parvum in stems | [81] |
| Bacillus subtilis PTA-271 | Rhizosphere soil | Significant reduction in full dieback by N. parvum in Chardonnay variety. Significant protective effect observed when co-applied with fungal BCA T. atroviride | [82] |
| Streptomyces sp. VV/E1 + VV/R4 | Root endophyte and rhizosphere soil | Very high biocontrol effect on black-foot disease pathogens Dactylonectria torresensis and Da. macrodidyma in 2-year field trials | [83] |
| P. oligandrum Po37 | Rhizosphere soil | Very high biocontrol effect on P. chlamydospora and P. minimum in 2-year field trials | [83] |
6.2. BCAs Based on Other Types of Bacteria
6.3. BCAs Based on Rhizospheric or Endophytic Fungi
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gramaje, D.; Úrbez-Torres, J.R.; Sosnowski, M.R.; Urbez-Torres, J.R.; Sosnowski, M.R. Managing Grapevine Trunk Diseases with Respect to Etiology and Epidemiology: Current Strategies and Future Prospects. Plant Dis. 2018, 102, 12–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramaje, D.; Armengol, J. Fungal Trunk Pathogens in the Grapevine Propagation Process: Potential Inoculum Sources, Detection, Identification, and Management Strategies. Plant Dis. 2011, 95, 1040–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agustí-Brisach, C.; Gramaje, D.; García-Jiménez, J.; Armengol, J. Detection of black-foot disease pathogens in the grapevine nursery propagation process in Spain. Eur. J. Plant Pathol. 2013, 137, 103–112. [Google Scholar] [CrossRef]
- Luque, J.; Elena, G.; Garcia-Figueres, F.; Reyes, J.; Barrios, G.; Legorburu, F.J. Natural infections of pruning wounds by fungal trunk pathogens in mature grapevines in Catalonia (Northeast Spain). Aust. J. Grape Wine Res. 2014, 20, 134–143. [Google Scholar] [CrossRef]
- Bertsch, C.; Ramírez-Suero, M.; Magnin-Robert, M.; Larignon, P.; Chong, J.; Abou-Mansour, E.; Spagnolo, A.; Clément, C.; Fontaine, F.; Clé, C.; et al. Grapevine trunk diseases: Complex and still poorly understood. Plant Pathol. 2013, 62, 243–265. [Google Scholar] [CrossRef] [Green Version]
- Aroca, Á.; Gramaje, D.; Armengol, J.; García-Jiménez, J.; Raposo, R. Evaluation of the grapevine nursery propagation process as a source of Phaeoacremonium spp. and Phaeomoniella chlamydospora and occurrence of trunk disease pathogens in rootstock mother vines in Spain. Eur. J. Plant Pathol. 2010, 126, 165–174. [Google Scholar] [CrossRef]
- Mondello, V.; Songy, A.; Battiston, E.; Pinto, C.; Coppin, C.; Trotel-Aziz, P.; Clément, C.; Mugnai, L.; Fontaine, F. Grapevine Trunk Diseases: A Review of Fifteen Years of Trials for Their Control with Chemicals and Biocontrol Agents. Plant Dis. 2018, 102, 1189–1217. [Google Scholar] [CrossRef] [Green Version]
- Bruisson, S.; Zufferey, M.; L’Haridon, F.; Trutmann, E.; Anand, A.; Dutartre, A.; De Vrieze, M.; Weisskopf, L. Endophytes and Epiphytes From the Grapevine Leaf Microbiome as Potential Biocontrol Agents against Phytopathogens. Front. Microbiol. 2019, 10, 2726. [Google Scholar] [CrossRef] [Green Version]
- Müller, D.B.; Vogel, C.; Bai, Y.; Vorholt, J.A. The Plant Microbiota: Systems-Level Insights and Perspectives. Annu. Rev. Genet. 2016, 50, 211–234. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, P.N.; Jha, D.K. Plant growth-promoting rhizobacteria (PGPR): Emergence in agriculture. World J. Microbiol. Biotechnol. 2012, 28, 1327–1350. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Pinto, C.; Pinho, D.; Sousa, S.; Pinheiro, M.; Egas, C.; Gomes, A.C. Unravelling the diversity of grapevine microbiome. PLoS ONE 2014, 9, e85622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarraonaindia, I.; Owens, S.M.; Weisenhorn, P.; West, K.; Hampton-Marcell, J.; Lax, S.; Bokulich, N.A.; Mills, D.A.; Martin, G.; Taghavi, S.; et al. The soil microbiome influences grapevine-associated microbiota. mBio 2015, 6, e02527-14. [Google Scholar] [CrossRef] [Green Version]
- Vionnet, L.; De Vrieze, M.; Dutartre, A.; Gfeller, A.; Lüthi, A.; L’Haridon, F.; Weisskopf, L. Microbial life in the grapevine: What can we expect from the leaf microbiome? Oeno One 2018, 52, 205–210. [Google Scholar] [CrossRef]
- Del Carmen Portillo, M.; Franquès, J.; Araque, I.; Reguant, C.; Bordons, A. Bacterial diversity of Grenache and Carignan grape surface from different vineyards at Priorat wine region (Catalonia, Spain). Int. J. Food Microbiol. 2016, 219, 56–63. [Google Scholar] [CrossRef]
- Salvetti, E.; Campanaro, S.; Campedelli, I.; Fracchetti, F.; Gobbi, A.; Tornielli, G.B.; Torriani, S.; Felis, G.E. Whole-metagenome-sequencing-based community profiles of Vitis vinifera L. cv. Corvina berries withered in two post-harvest conditions. Front. Microbiol. 2016, 7, 937. [Google Scholar] [CrossRef] [Green Version]
- Burns, K.N.; Bokulich, N.A.; Cantu, D.; Greenhut, R.F.; Kluepfel, D.A.; O’Geen, A.T.; Strauss, S.L.; Steenwerth, K.L. Vineyard soil bacterial diversity and composition revealed by 16S rRNA genes: Differentiation by vineyard management. Soil Biol. Biochem. 2016, 103, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Castañeda, L.E.; Barbosa, O. Metagenomic analysis exploring taxonomic and functional diversity of soil microbial communities in Chilean vineyards and surrounding native forests. PeerJ 2017, 5, e3098. [Google Scholar] [CrossRef]
- Longa, C.M.O.; Nicola, L.; Antonielli, L.; Mescalchin, E.; Zanzotti, R.; Turco, E.; Pertot, I. Soil microbiota respond to green manure in organic vineyards. J. Appl. Microbiol. 2017, 123, 1547–1560. [Google Scholar] [CrossRef]
- Novello, G.; Gamalero, E.; Bona, E.; Boatti, L.; Mignone, F.; Massa, N.; Cesaro, P.; Lingua, G.; Berta, G. The rhizosphere bacterial microbiota of Vitis vinifera cv. Pinot Noir in an integrated pest management vineyard. Front. Microbiol. 2017, 8, 1528. [Google Scholar] [CrossRef]
- Samad, A.; Trognitz, F.; Compant, S.; Antonielli, L.; Sessitsch, A. Shared and host-specific microbiome diversity and functioning of grapevine and accompanying weed plants. Environ. Microbiol. 2017, 19, 1407–1424. [Google Scholar] [CrossRef] [PubMed]
- Chou, M.Y.; Vanden Heuvel, J.; Bell, T.H.; Panke-Buisse, K.; Kao-Kniffin, J. Vineyard under-vine floor management alters soil microbial composition, while the fruit microbiome shows no corresponding shifts. Sci. Rep. 2018, 8, 11039. [Google Scholar] [CrossRef]
- D’Amico, F.; Candela, M.; Turroni, S.; Biagi, E.; Brigidi, P.; Bega, A.; Vancini, D.; Rampelli, S. The rootstock regulates microbiome diversity in root and rhizosphere compartments of Vitis vinifera cultivar lambrusco. Front. Microbiol. 2018, 9, 2240. [Google Scholar] [CrossRef] [PubMed]
- Hendgen, M.; Hoppe, B.; Döring, J.; Friedel, M.; Kauer, R.; Frisch, M.; Dahl, A.; Kellner, H. Effects of different management regimes on microbial biodiversity in vineyard soils. Sci. Rep. 2018, 8, 9393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayawardena, R.S.; Purahong, W.; Zhang, W.; Wubet, T.; Li, X.; Liu, M.; Zhao, W.; Hyde, K.D.; Liu, J.; Yan, J. Biodiversity of fungi on Vitis vinifera L. revealed by traditional and high-resolution culture-independent approaches. Fungal Divers. 2018, 90, 1–84. [Google Scholar] [CrossRef] [Green Version]
- Dissanayake, A.J.; Purahong, W.; Wubet, T.; Hyde, K.D.; Zhang, W.; Xu, H.; Zhang, G.; Fu, C.; Liu, M.; Xing, Q.; et al. Direct comparison of culture-dependent and culture-independent molecular approaches reveal the diversity of fungal endophytic communities in stems of grapevine (Vitis vinifera). Fungal Divers. 2018, 90, 85–107. [Google Scholar] [CrossRef]
- Morales-Cruz, A.; Allenbeck, G.; Figueroa-Balderas, R.; Ashworth, V.E.; Lawrence, D.P.; Travadon, R.; Smith, R.J.; Baumgartner, K.; Rolshausen, P.E.; Cantu, D. Closed-reference metatranscriptomics enables in planta profiling of putative virulence activities in the grapevine trunk disease complex. Mol. Plant Pathol. 2018, 19, 490–503. [Google Scholar] [CrossRef] [Green Version]
- Eichmeier, A.; Pečenka, J.; Peňázová, E.; Baránek, M.; Català-García, S.; León, M.; Armengol, J.; Gramaje, D. High-throughput amplicon sequencing-based analysis of active fungal communities inhabiting grapevine after hot-water treatments reveals unexpectedly high fungal diversity. Fungal Ecol. 2018, 36, 26–38. [Google Scholar] [CrossRef]
- Marasco, R.; Rolli, E.; Fusi, M.; Michoud, G.; Daffonchio, D. Grapevine rootstocks shape underground bacterial microbiome and networking but not potential functionality. Microbiome 2018, 6, 3. [Google Scholar] [CrossRef]
- Del Frari, G.; Gobbi, A.; Aggerbeck, M.R.; Oliveira, H.; Hansen, L.H.; Ferreira, R.B. Characterization of the Wood Mycobiome of Vitis vinifera in a Vineyard Affected by Esca. Spatial Distribution of Fungal Communities and Their Putative Relation with Leaf Symptoms. Front. Plant Sci. 2019, 10, 910. [Google Scholar] [CrossRef] [Green Version]
- Berlanas, C.; Berbegal, M.; Elena, G.; Laidani, M.; Cibriain, J.F.; Sagües, A.; Gramaje, D. The fungal and bacterial rhizosphere microbiome associated with grapevine rootstock genotypes in mature and young vineyards. Front. Microbiol. 2019, 10, 1142. [Google Scholar] [CrossRef] [PubMed]
- Del Pilar Martínez-Diz, M.; Díaz-Losada, E.; Barajas, E.; Ruano-Rosa, D.; Andrés-Sodupe, M.; Gramaje, D. Screening of Spanish Vitis vinifera germplasm for resistance to Phaeomoniella chlamydospora. Sci. Hortic. 2019, 246, 104–109. [Google Scholar] [CrossRef]
- Niem, J.M.; Billones-Baaijens, R.; Stodart, B.; Savocchia, S. Diversity Profiling of Grapevine Microbial Endosphere and Antagonistic Potential of Endophytic Pseudomonas against Grapevine Trunk Diseases. Front. Microbiol. 2020, 11, 477. [Google Scholar] [CrossRef] [PubMed]
- Dries, L.; Bussotti, S.; Pozzi, C.; Kunz, R.; Schnell, S.; Löhnertz, O.; Vortkamp, A. Rootstocks shape their microbiome—Bacterial communities in the rhizosphere of different grapevine rootstocks. Microorganisms 2021, 9, 822. [Google Scholar] [CrossRef] [PubMed]
- Deyett, E.; Rolshausen, P.E. Endophytic microbial assemblage in grapevine. FEMS Microbiol. Ecol. 2020, 96, fiaa053. [Google Scholar] [CrossRef]
- Fotios, B.; Sotirios, V.; Elena, P.; Anastasios, S.; Stefanos, T.; Danae, G.; Georgia, T.; Aliki, T.; Epaminondas, P.; Emmanuel, M.; et al. Grapevine wood microbiome analysis identifies key fungal pathogens and potential interactions with the bacterial community implicated in grapevine trunk disease appearance. Environ. Microbiome 2021, 16, 23. [Google Scholar] [CrossRef]
- Paolinelli, M.; Escoriaza, G.; Cesari, C.; Garcia-Lampasona, S.; Hernandez-Martinez, R. Characterization of Grapevine Wood Microbiome through a Metatranscriptomic Approach. Microb. Ecol. 2021. [Google Scholar] [CrossRef]
- Lade, S.B.; Štraus, D.; Oliva, J. Variation in Fungal Community in Grapevine (Vitis vinifera) Nursery Stock Depends on Nursery, Variety and Rootstock. J. Fungi 2022, 8, 47. [Google Scholar] [CrossRef]
- Jayawardena, R.S.; Liu, M.; Maharachchikumbura, S.S.N.; Zhang, W.; Xing, Q.; Hyde, K.D.; Nilthong, S.; Li, X.; Yan, J. Neopestalotiopsis vitis sp. Nov. causing grapevine leaf spot in China. Phytotaxa 2016, 258, 63–74. [Google Scholar] [CrossRef]
- Dissanayake, A.J.; Liu, M.; Zhang, W.; Chen, Z.; Udayanga, D.; Chukeatirote, E.; Li, X.H.; Yan, J.Y.; Hyde, K.D. Morphological and molecular characterisation of Diaporthe species associated with grapevine trunk disease in China. Fungal Biol. 2015, 119, 283–294. [Google Scholar] [CrossRef]
- González, V.; Sánchez-Torres, P.; Hinarejos, R.; Tuset, J.J. Inonotus hispidus fruiting bodies on grapevines with esca symptoms in mediterranean areas of Spain. J. Plant Pathol. 2009, 91, 465–468. [Google Scholar]
- Massonnet, M.; Morales-Cruz, A.; Minio, A.; Figueroa-Balderas, R.; Lawrence, D.P.; Travadon, R.; Rolshausen, P.E.; Baumgartner, K.; Cantu, D. Whole-genome resequencing and pan-transcriptome reconstruction highlight the impact of genomic structural variation on secondary metabolite gene clusters in the grapevine esca pathogen Phaeoacremonium minimum. Front. Microbiol. 2018, 9, 1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatica, M.; Dubos, B.; Larignon, P. The “hoja de malvón” grape disease in Argentina. Phytopathol. Mediterr. 2000, 39, 41–45. [Google Scholar] [CrossRef]
- Egamberdieva, D.; Kamilova, F.; Validov, S.; Gafurova, L.; Kucharova, Z.; Lugtenberg, B. High incidence of plant growth-stimulating bacteria associated with the rhizosphere of wheat grown on salinated soil in Uzbekistan. Environ. Microbiol. 2008, 10, 1–9. [Google Scholar] [CrossRef]
- Mendes, R.; Kruijt, M.; De Bruijn, I.; Dekkers, E.; van der Voort, M.; Schneider, J.H.M.M.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.H.M.H.M.; et al. Deciphering the Rhizosphere Microbiome for Disease-Suppressive Bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef]
- Marilley, L.; Aragno, M. Phylogenetic diversity of bacterial communities differing in degree of proximity of Lolium perenne and Trifolium repens roots. Appl. Soil Ecol. 1999, 13, 127–136. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; Ver Loren van Themaat, E.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef] [Green Version]
- Quiza, L.; St-Arnaud, M.; Yergeau, E. Harnessing phytomicrobiome signaling for rhizosphere microbiome engineering. Front. Plant Sci. 2015, 6, 507. [Google Scholar] [CrossRef]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; Van Der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef]
- Bazghaleh, N.; Hamel, C.; Gan, Y.; Tar’an, B.; Knight, J.D. Genotype-Specific Variation in the Structure of Root Fungal Communities Is Related to Chickpea Plant Productivity. Appl. Environ. Microbiol. 2015, 81, 2368–2377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Thorngate, J.H.; Richardson, P.M.; Mills, D.A. Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. USA 2014, 111, E139–E148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, K.N.; Kluepfel, D.A.; Strauss, S.L.; Bokulich, N.A.; Cantu, D.; Steenwerth, K.L. Vineyard soil bacterial diversity and composition revealed by 16S rRNA genes: Differentiation by geographic features. Soil Biol. Biochem. 2015, 91, 232–247. [Google Scholar] [CrossRef] [Green Version]
- DeBruyn, J.M.; Nixon, L.T.; Fawaz, M.N.; Johnson, A.M.; Radosevich, M. Global biogeography and quantitative seasonal dynamics of Gemmatimonadetes in soil. Appl. Environ. Microbiol. 2011, 77, 6295–6300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warschefsky, E.J.; Klein, L.L.; Frank, M.H.; Chitwood, D.H.; Londo, J.P.; von Wettberg, E.J.B.; Miller, A.J. Rootstocks: Diversity, Domestication, and Impacts on Shoot Phenotypes. Trends Plant Sci. 2016, 21, 418–437. [Google Scholar] [CrossRef]
- Habran, A.; Commisso, M.; Helwi, P.; Hilbert, G.; Negri, S.; Ollat, N.; Gomès, E.; Van Leeuwen, C.; Guzzo, F.; Delrot, S. Roostocks/scion/nitrogen interactions affect secondary metabolism in the grape berry. Front. Plant Sci. 2016, 7, 1134. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, S.N.; Waite, I.S.; Blackburn, A.; Husband, R.; Rushton, S.P.; Manning, D.C.; O’Donnell, A.G. Actinobacterial community dynamics in long term managed grasslands. Antonie Van Leeuwenhoek 2009, 95, 319–334. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Fierer, N.; Schimel, J.P.; Holden, P.A. Variations in microbial community composition through two soil depth profiles. Soil Biol. Biochem. 2003, 35, 167–176. [Google Scholar] [CrossRef]
- Kielak, A.M.; Barreto, C.C.; Kowalchuk, G.A.; van Veen, J.A.; Kuramae, E.E. The Ecology of Acidobacteria: Moving beyond Genes and Genomes. Front. Microbiol. 2016, 7, 744. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, E.; Zilber-Rosenberg, I. Microbes Drive Evolution of Animals and Plants: The Hologenome Concept. mBio 2016, 7, e01395-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarrete, A.A.; Kuramae, E.E.; de Hollander, M.; Pijl, A.S.; van Veen, J.A.; Tsai, S.M. Acidobacterial community responses to agricultural management of soybean in Amazon forest soils. FEMS Microbiol. Ecol. 2013, 83, 607–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramaje, D.; García-Jiménez, J.; Armengol, J. Field Evaluation of Grapevine Rootstocks Inoculated with Fungi Associated with Petri Disease and Esca. Am. J. Enol. Vitic. 2010, 61, 512–520. [Google Scholar] [CrossRef]
- Deketelaere, S.; Tyvaert, L.; França, S.C.; Hofte, M. Desirable traits of a good biocontrol agent against Verticillium wilt. Front. Microbiol. 2017, 8, 1186. [Google Scholar] [CrossRef] [Green Version]
- Rolshausen, P.E.; Úrbez-Torres, J.R.; Rooney-Latham, S.; Eskalen, A.; Smith, R.J.; Gubler, W.D. Evaluation of Pruning Wound Susceptibility and Protection against Fungi Associated with Grapevine Trunk Diseases. Am. J. Enol. Vitic. 2010, 61, 113–119. [Google Scholar]
- Van Niekerk, J.M.; Halleen, F.; Fourie, P.H. Temporal susceptibility of grapevine pruning wounds to trunk pathogen infection in South African grapevines. Phytopathol. Mediterr. 2011, 50, 139–150. [Google Scholar] [CrossRef]
- Úrbez-Torres, J.R.; Gubler, W.D. Susceptibility of grapevine pruning wounds to infection by Lasiodiplodia theobromae and Neofusicoccum parvum. Plant Pathol. 2011, 60, 261–270. [Google Scholar] [CrossRef]
- Coque, J.J.R.; Álvarez-Pérez, J.M.; Cobos, R.; González-García, S.; Ibáñez, A.M.; Diez Galán, A.; Calvo-Peña, C. Advances in the control of phytopathogenic fungi that infect crops through their root system. In Advances in Applied Microbiology; Geoffrey Michael Gadd, S.S., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 111, pp. 123–170. ISBN 9780128207055. [Google Scholar]
- Hofstetter, V.; Buyck, B.; Croll, D.; Viret, O.; Couloux, A.; Gindro, K. What if esca disease of grapevine were not a fungal disease? Fungal Divers. 2012, 54, 51–67. [Google Scholar] [CrossRef] [Green Version]
- Janssen, P.H. Identifying the Dominant Soil Bacterial Taxa in Libraries of 16S rRNA and 16S rRNA Genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef] [Green Version]
- Doumbou, C.L.; Hamby Salove, M.K.; Crawford, D.L.; Beaulieu, C. Actinomycetes, promising tools to control plant diseases and to promote plant growth. Phytoprotection 2001, 82, 85–102. [Google Scholar] [CrossRef] [Green Version]
- Davelos, A.L.; Kinkel, L.L.; Samac, D.A. Spatial Variation in Frequency and Intensity of Antibiotic Interactions among Streptomycetes from Prairie Soil. Appl. Environ. Microbiol. 2004, 70, 1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrey, S.D.; Tarkka, M.T. Friends and foes: Streptomycetes as modulators of plant disease and symbiosis. Antonie Van Leeuwenhoek 2008, 94, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Labeda, D.P.; Goodfellow, M.; Brown, R.; Ward, A.C.; Lanoot, B.; Vanncanneyt, M.; Swings, J.; Kim, S.-B.; Liu, Z.; Chun, J.; et al. Phylogenetic study of the species within the family Streptomycetaceae. Antonie Van Leeuwenhoek 2012, 101, 73–104. [Google Scholar] [CrossRef] [PubMed]
- Van der Meij, A.; Worsley, S.F.; Hutchings, M.I.; van Wezel, G.P. Chemical ecology of antibiotic production by actinomycetes. FEMS Microbiol. Rev. 2017, 41, 392–416. [Google Scholar] [CrossRef]
- Vurukonda, S.S.K.P.; Giovanardi, D.; Stefani, E. Plant growth promoting and biocontrol activity of Streptomyces spp. As endophytes. Int. J. Mol. Sci. 2018, 19, 952. [Google Scholar] [CrossRef] [Green Version]
- Álvarez-Pérez, J.M.; González-García, S.; Cobos, R.; Olego, M.Á.; Ibañez, A.; Díez-Galán, A.; Garzón-Jimeno, E.; Coque, J.J.R. Use of endophytic and rhizosphere actinobacteria from grapevine plants to reduce nursery fungal graft infections that lead to young grapevine decline. Appl. Environ. Microbiol. 2017, 83, e01564-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haidar, R.; Roudet, J.; Bonnard, O.; Dufour, M.C.; Corio-Costet, M.F.; Fert, M.; Gautier, T.; Deschamps, A.; Fermaud, M. Screening and modes of action of antagonistic bacteria to control the fungal pathogen Phaeomoniella chlamydospora involved in grapevine trunk diseases. Microbiol. Res. 2016, 192, 172–184. [Google Scholar] [CrossRef]
- Yacoub, A.; Gerbore, J.; Magnin, N.; Chambon, P.; Dufour, M.C.; Corio-Costet, M.F.; Guyoneaud, R.; Rey, P. Ability of Pythium oligandrum strains to protect Vitis vinifera L., by inducing plant resistance against Phaeomoniella chlamydospora, a pathogen involved in Esca, a grapevine trunk disease. Biol. Control 2016, 92, 7–16. [Google Scholar] [CrossRef]
- Daraignes, L.; Gerbore, J.; Yacoub, A.; Dubois, L.; Romand, C.; Zekri, O.; Roudet, J.; Chambon, P.; Fermaud, M. Efficacy of P. oligandrum affected by its association with bacterial BCAs and rootstock effect in controlling grapevine trunk diseases. Biol. Control 2018, 119, 59–67. [Google Scholar] [CrossRef]
- Haidar, R.; Amira, Y.; Roudet, J.; Marc, F.; Patrice, R. Application methods and modes of action of Pantoea agglomerans and Paenibacillus sp. to control the grapevine trunk disease-pathogen, Neofusicoccum parvum. OENO One 2021, 55, 1–16. [Google Scholar] [CrossRef]
- Leal, C.; Richet, N.; Guise, J.; Gramaje, D. Cultivar Contributes to the Beneficial Effects of Bacillus subtilis PTA-271 and Trichoderma atroviride SC1 to Protect Grapevine against Neofusicoccum parvum. Front. Microbiol. 2021, 12, 2889. [Google Scholar] [CrossRef] [PubMed]
- Pilar Martínez-Diz, M.; Díaz-Losada, E.; Andrés-Sodupe, M.; Bujanda, R.; Maldonado-González, M.M.; Ojeda, S.; Yacoub, A.; Rey, P.; Gramaje, D. Field evaluation of biocontrol agents against black-foot and Petri diseases of grapevine. Pest Manag. Sci. 2021, 77, 697–708. [Google Scholar] [CrossRef] [PubMed]
- González-García, S.; Álvarez-Pérez, J.M.; Sáenz de Miera, L.E.; Cobos, R.; Ibañez, A.; Díez-Galán, A.; Garzón-Jimeno, E.; Coque, J.J.R. Developing tools for evaluating inoculation methods of biocontrol Streptomyces sp. strains into grapevine plants. PLoS ONE 2019, 14, e0211225. [Google Scholar] [CrossRef] [Green Version]
- Andreolli, M.; Lampis, S.; Zapparoli, G.; Angelini, E.; Vallini, G. Diversity of bacterial endophytes in 3 and 15 year-old grapevines of Vitis vinifera cv. Corvina and their potential for plant growth promotion and phytopathogen control. Microbiol. Res. 2016, 183, 42–52. [Google Scholar] [CrossRef]
- Haidar, R.; Deschamps, A.; Roudet, J.; Calvo-Garrido, C.; Bruez, E.; Rey, P.; Fermaud, M. Multi-organ screening of efficient bacterial control agents against two major pathogens of grapevine. Biol. Control 2016, 92, 55–65. [Google Scholar] [CrossRef]
- Russi, A.; Almança, M.A.K.; Grohs, D.S.; Schwambach, J. Biocontrol of black foot disease on grapevine rootstocks using Bacillus subtilis strain F62. Trop. Plant Pathol. 2020, 45, 103–111. [Google Scholar] [CrossRef]
- Pertot, I.; Prodorutti, D.; Colombini, A.; Pasini, L. Trichoderma atroviride SC1 prevents Phaeomoniella chlamydospora and Phaeoacremonium aleophilum infection of grapevine plants during the grafting process in nurseries. BioControl 2016, 61, 257–267. [Google Scholar] [CrossRef]
- Silva-Valderrama, I.; Toapanta, D.; Miccono, M.D.; Lolas, M.; Díaz, G.A.; Cantu, D.; Castro, A. Biocontrol Potential of Grapevine Endophytic and Rhizospheric Fungi against Trunk Pathogens. Front. Microbiol. 2021, 11, 3311. [Google Scholar] [CrossRef]
- Billar de Almeida, A.; Concas, J.; Campos, M.D.; Materatski, P.; Varanda, C.; Patanita, M.; Murolo, S.; Romanazzi, G.; Félix, M.D.R. Endophytic Fungi as Potential Biological Control Agents against Grapevine Trunk Diseases in Alentejo Region. Biology 2020, 9, 420. [Google Scholar] [CrossRef]
- Gerbore, J.; Vallance, J.; Yacoub, A.; Delmotte, F.; Grizard, D.; Regnault-Roger, C.; Rey, P. Characterization of Pythium oligandrum populations that colonize the rhizosphere of vines from the Bordeaux region. FEMS Microbiol. Ecol. 2014, 90, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Yacoub, A.; Magnin, N.; Gerbore, J.; Haidar, R.; Bruez, E.; Compant, S.; Guyoneaud, R.; Rey, P. The biocontrol root-oomycete, Pythium oligandrum, triggers grapevine resistance and shifts in the transcriptome of the trunk pathogenic fungus, Phaeomoniella chlamydospora. Int. J. Mol. Sci. 2020, 21, 6876. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cobos, R.; Ibañez, A.; Diez-Galán, A.; Calvo-Peña, C.; Ghoreshizadeh, S.; Coque, J.J.R. The Grapevine Microbiome to the Rescue: Implications for the Biocontrol of Trunk Diseases. Plants 2022, 11, 840. https://doi.org/10.3390/plants11070840
Cobos R, Ibañez A, Diez-Galán A, Calvo-Peña C, Ghoreshizadeh S, Coque JJR. The Grapevine Microbiome to the Rescue: Implications for the Biocontrol of Trunk Diseases. Plants. 2022; 11(7):840. https://doi.org/10.3390/plants11070840
Chicago/Turabian StyleCobos, Rebeca, Ana Ibañez, Alba Diez-Galán, Carla Calvo-Peña, Seyedehtannaz Ghoreshizadeh, and Juan José R. Coque. 2022. "The Grapevine Microbiome to the Rescue: Implications for the Biocontrol of Trunk Diseases" Plants 11, no. 7: 840. https://doi.org/10.3390/plants11070840
APA StyleCobos, R., Ibañez, A., Diez-Galán, A., Calvo-Peña, C., Ghoreshizadeh, S., & Coque, J. J. R. (2022). The Grapevine Microbiome to the Rescue: Implications for the Biocontrol of Trunk Diseases. Plants, 11(7), 840. https://doi.org/10.3390/plants11070840