
Genetic Diversity, Population Structure and Linkage Disequilibrium Analyses in Tropical Maize Using Genotyping by Sequencing

 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Results
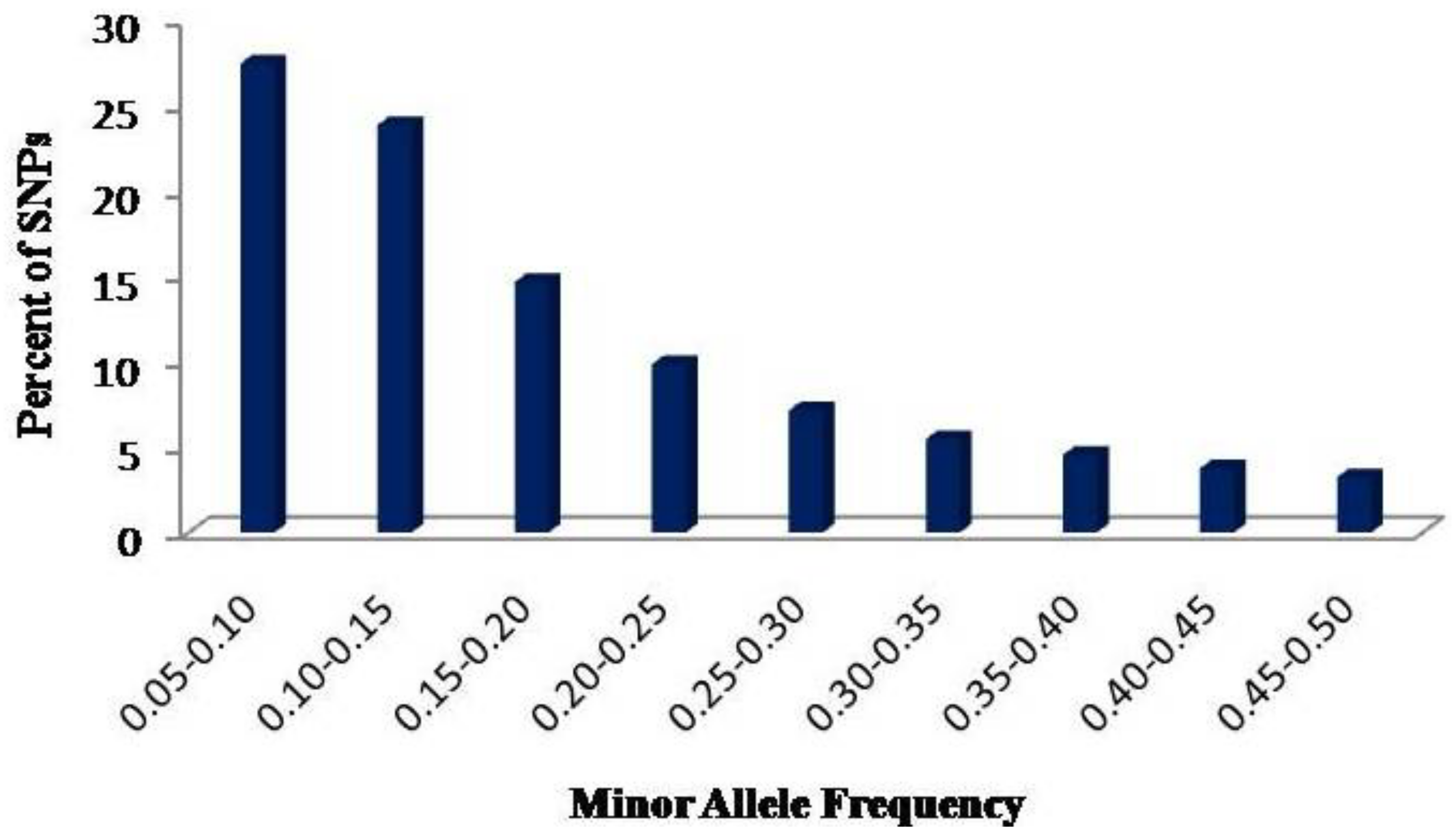
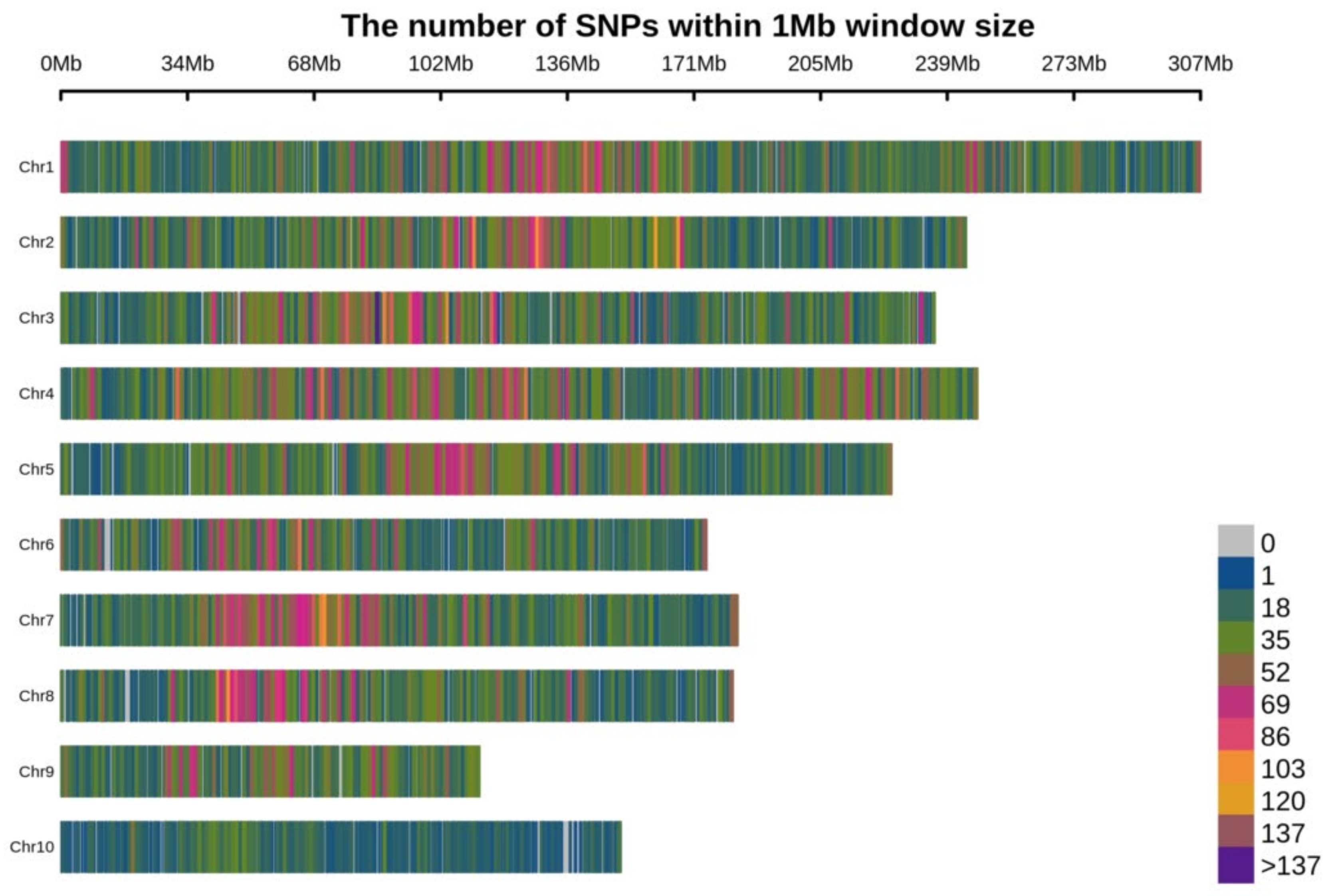
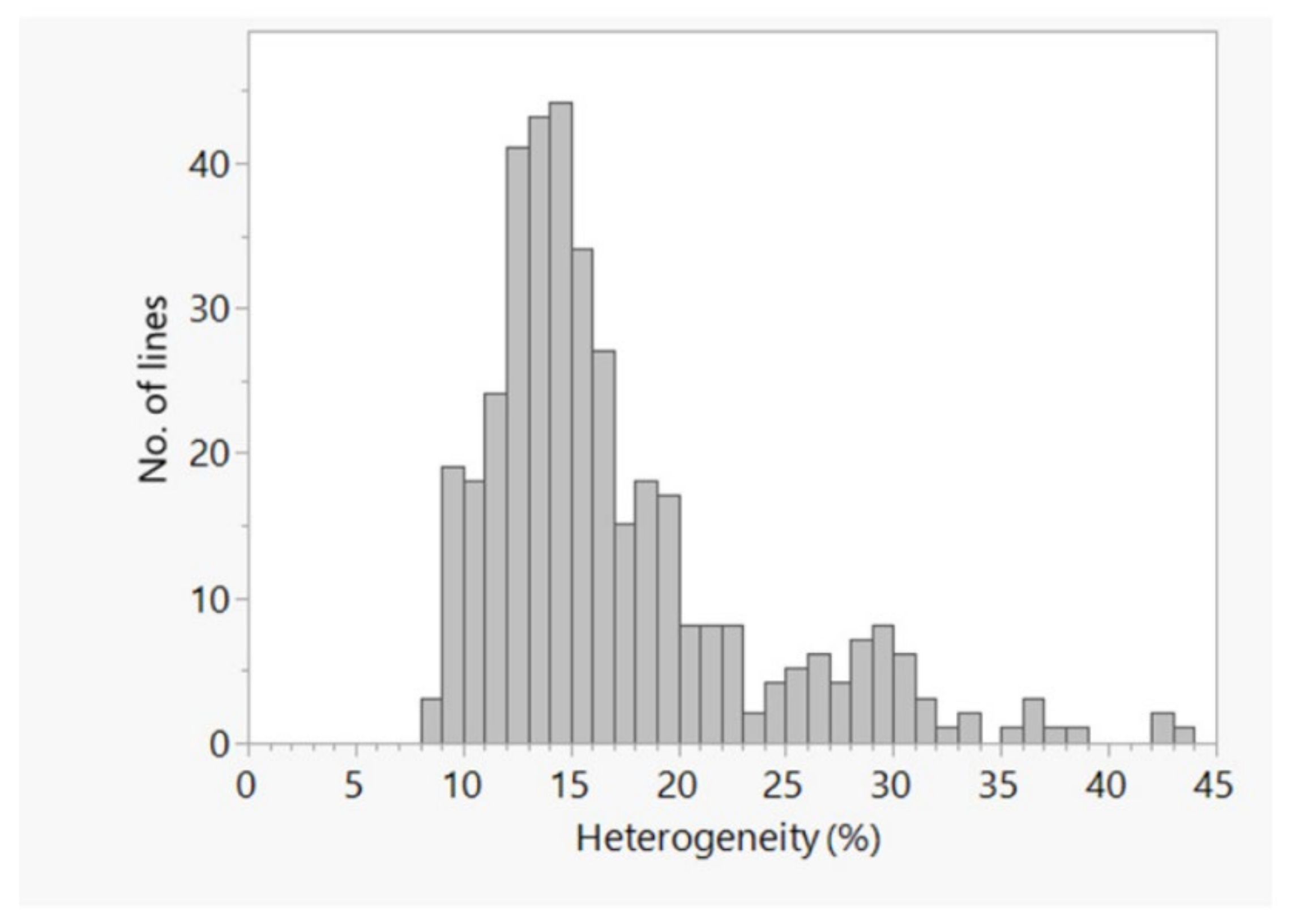
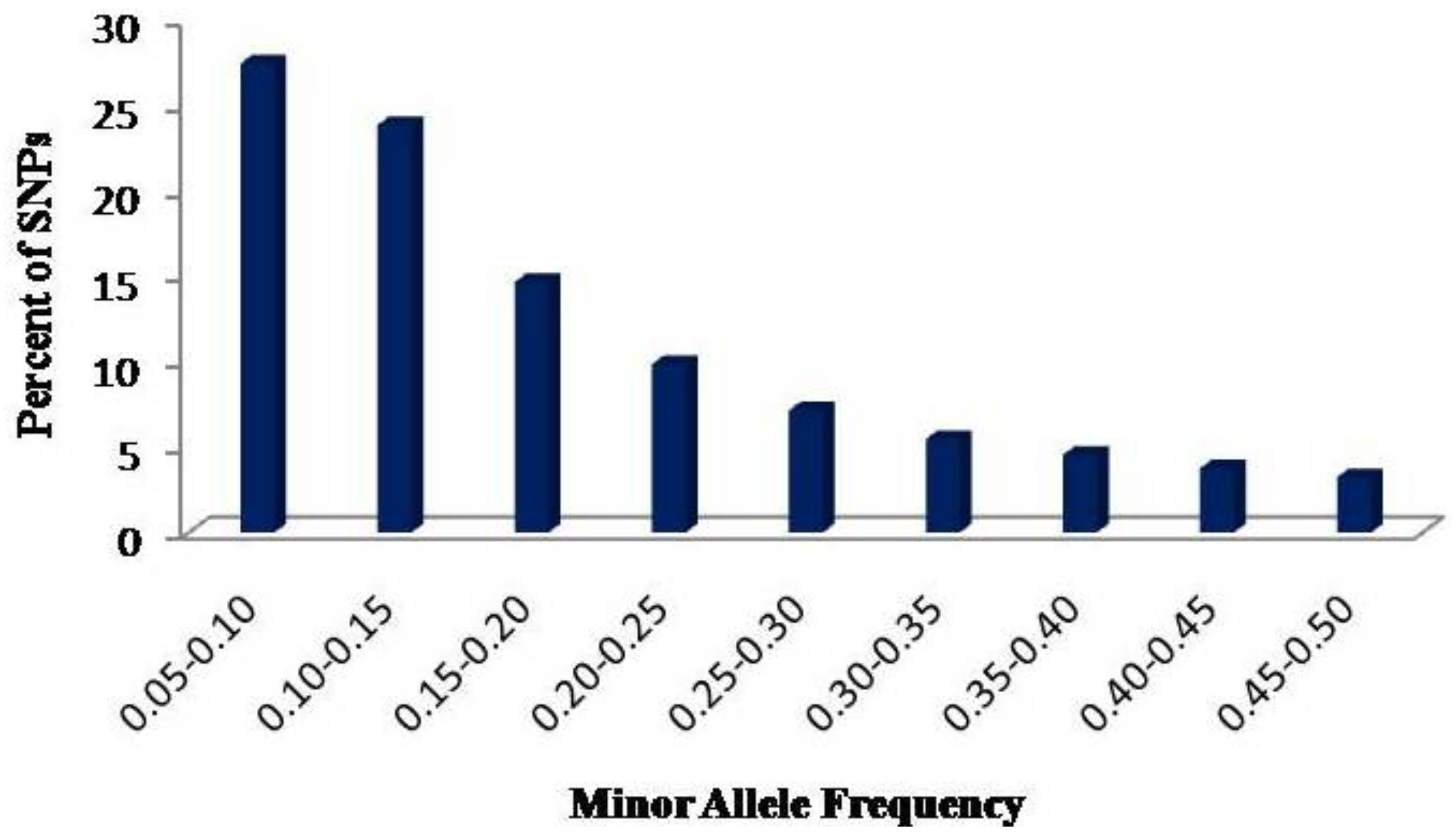
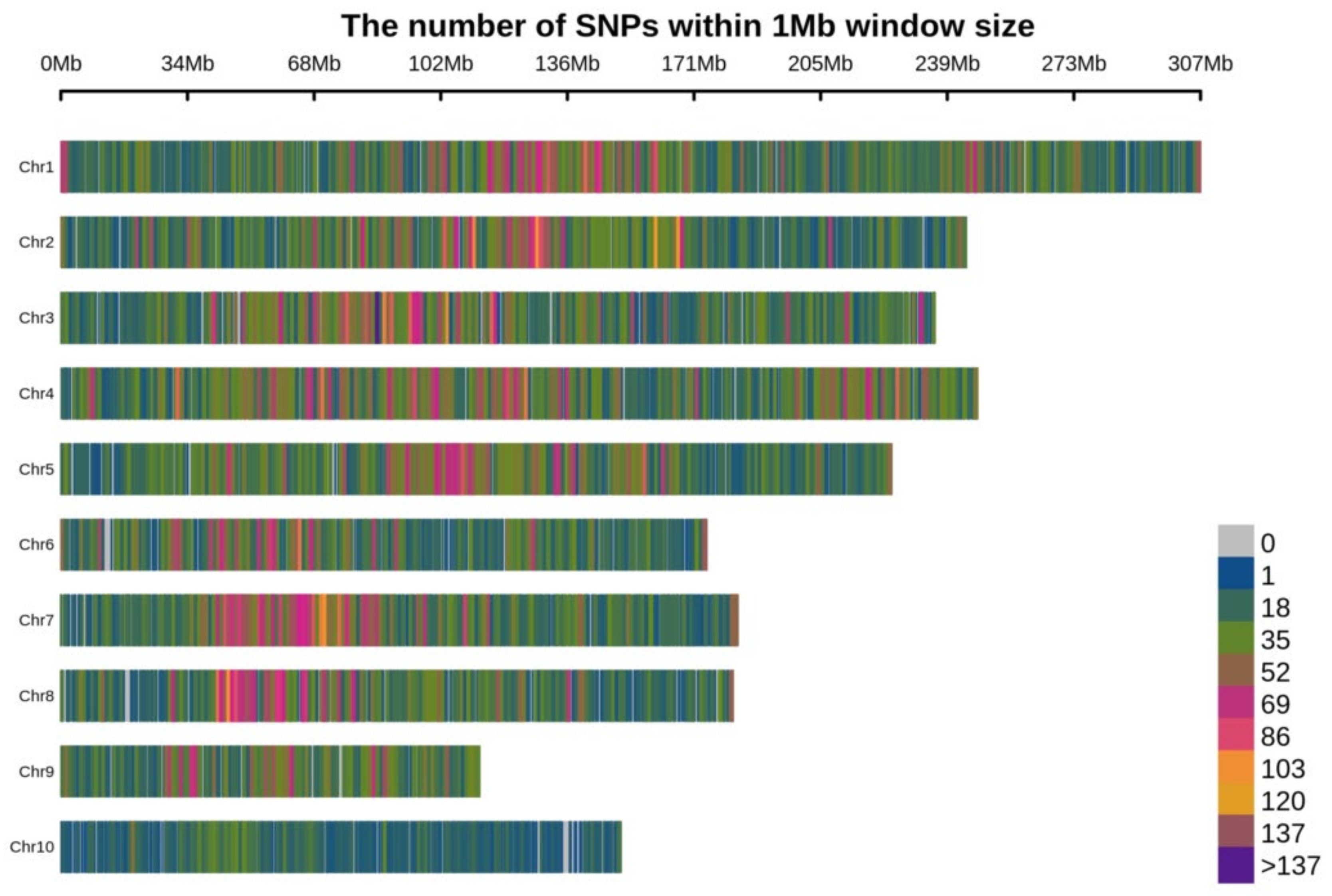
2.1. SNP Discovery and Their Distribution
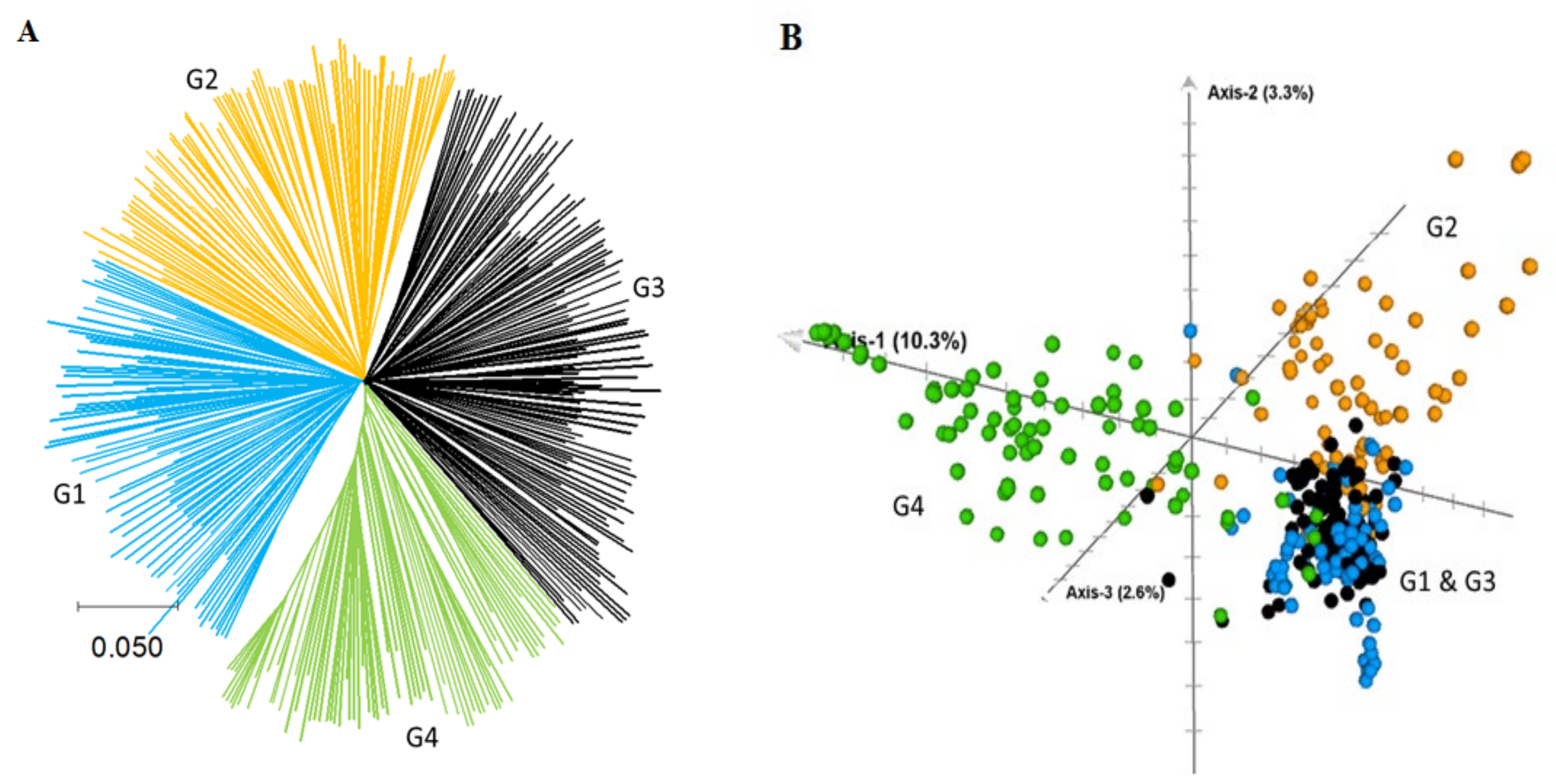
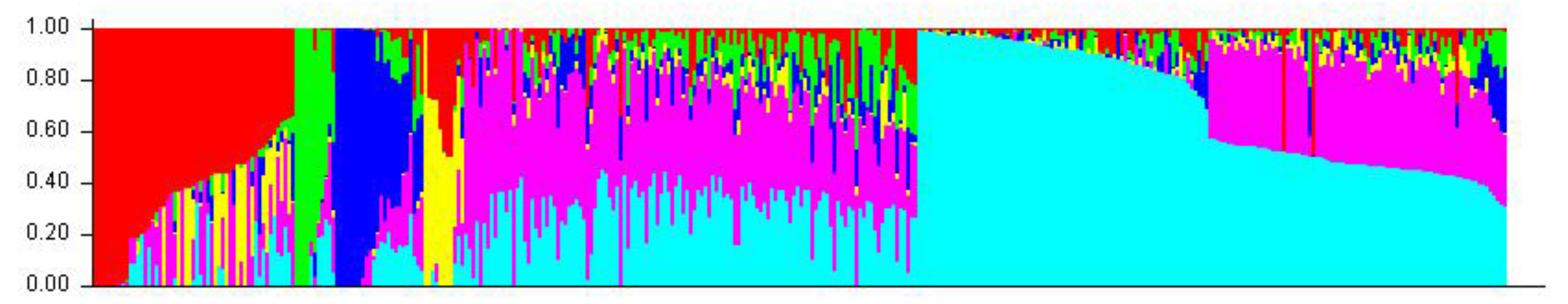
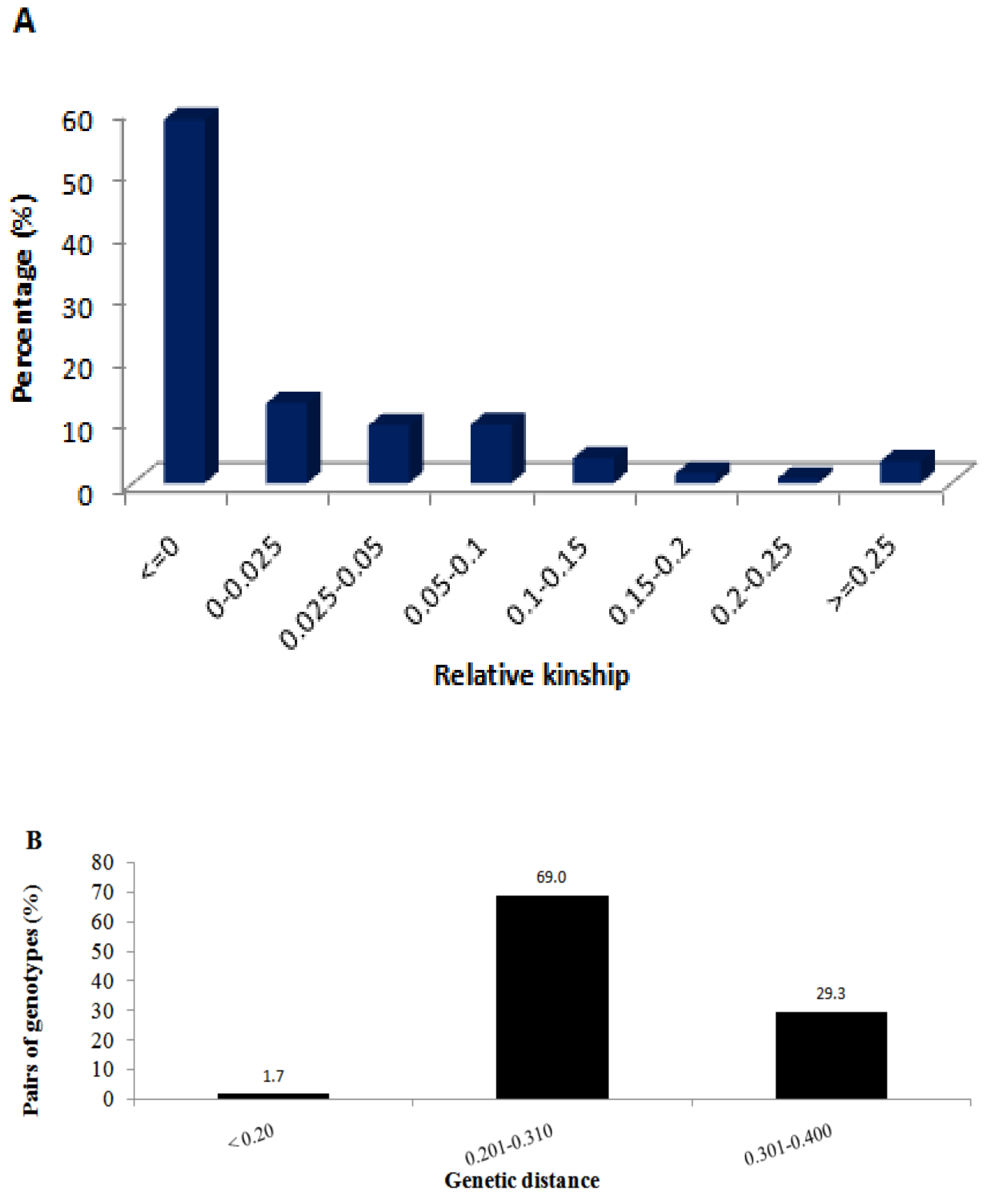
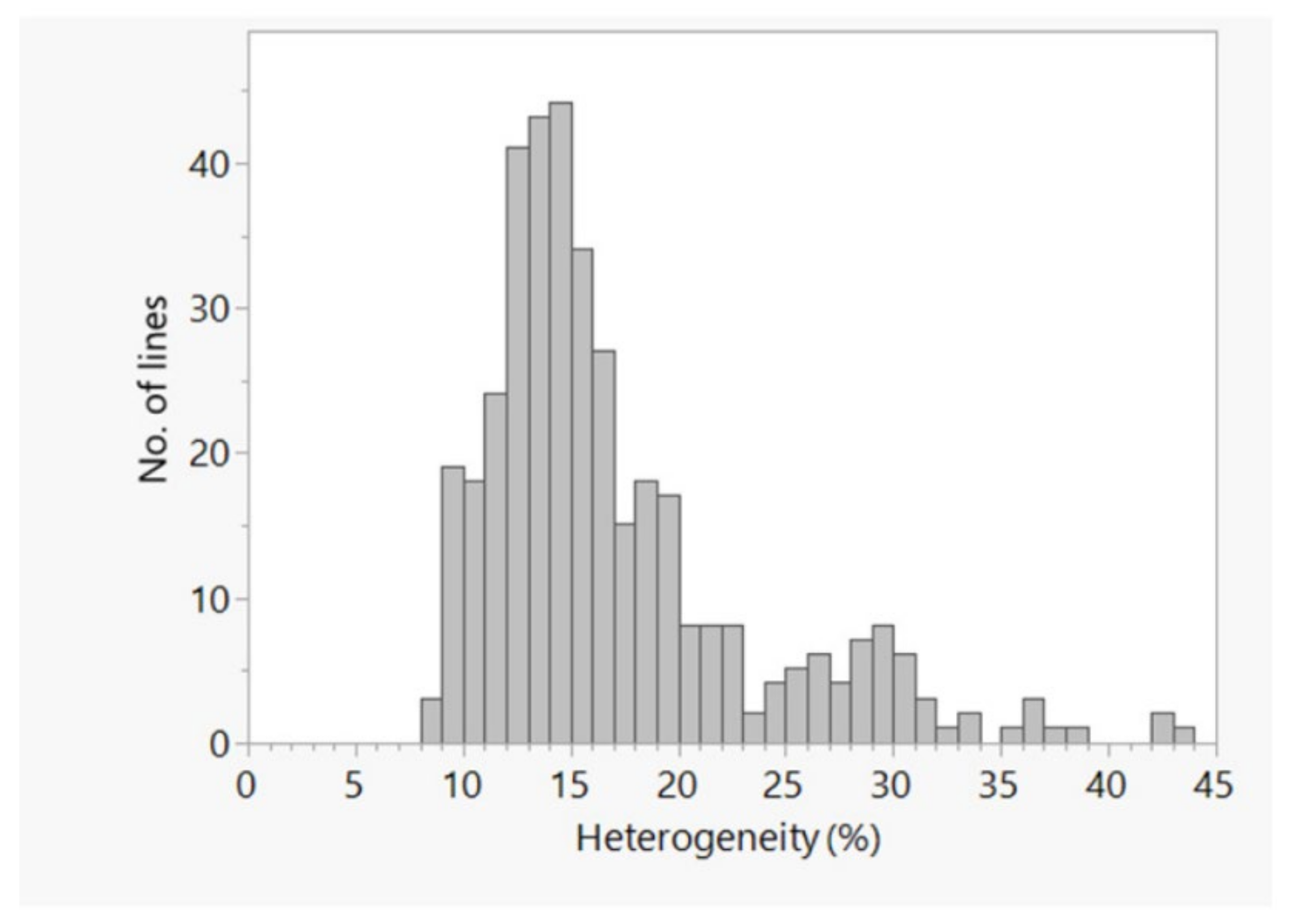
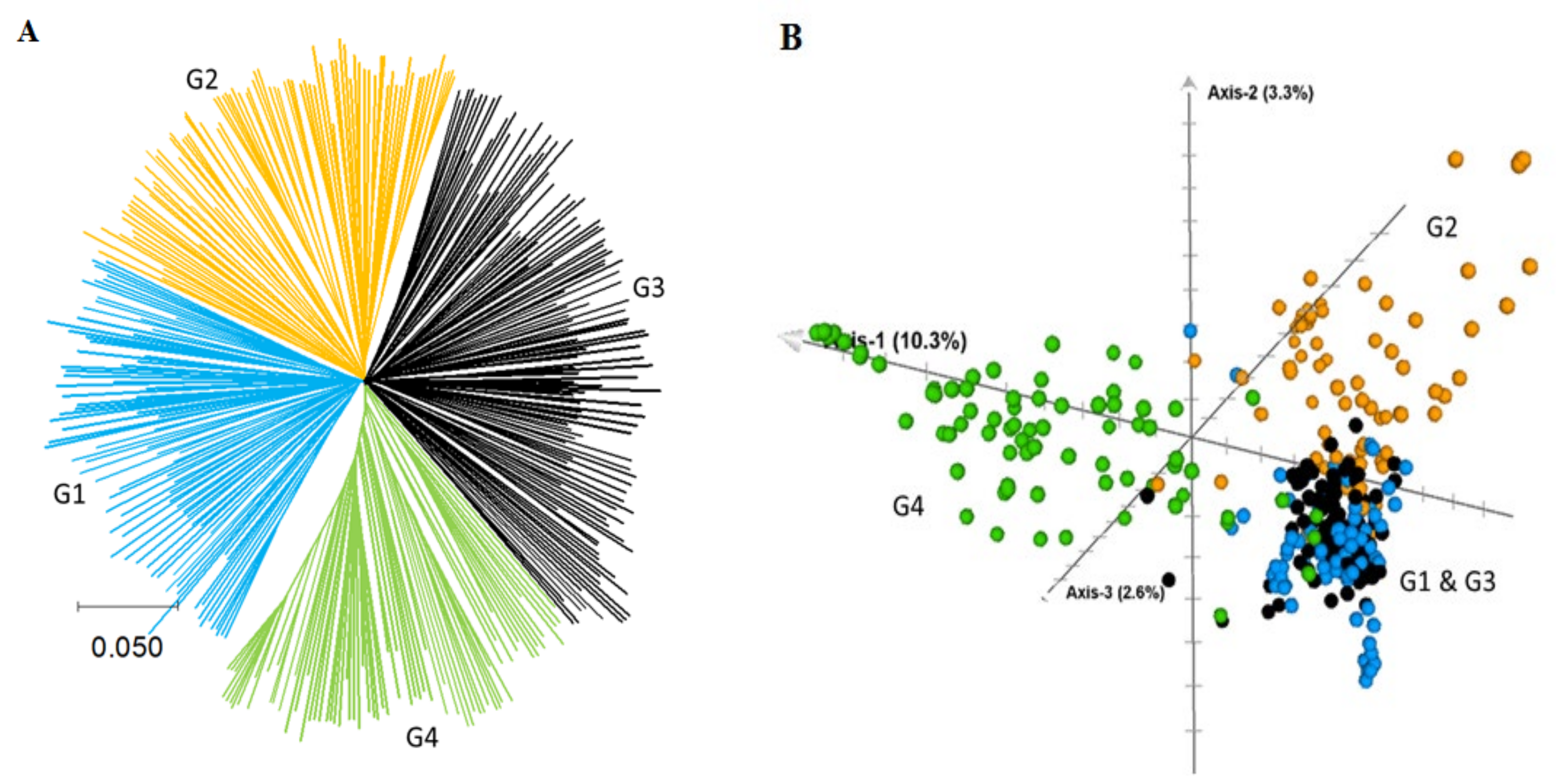
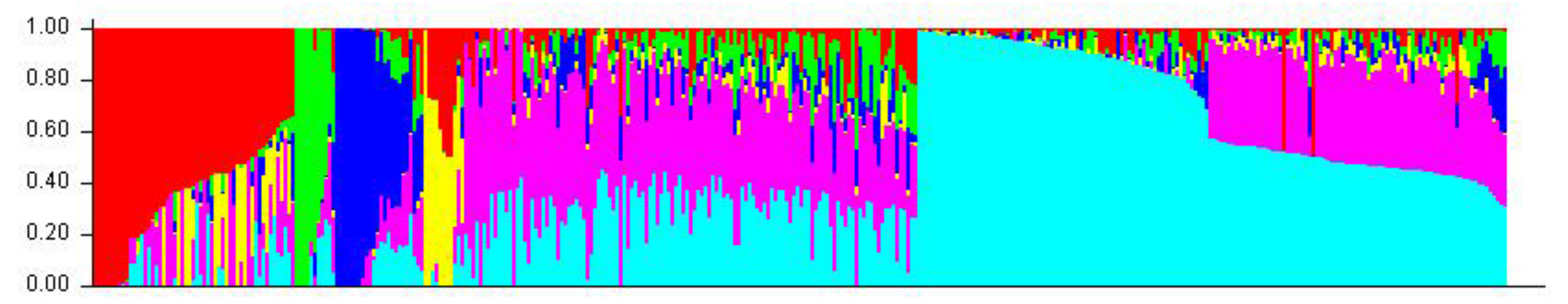
2.2. Genetic Relationship and Population Structure
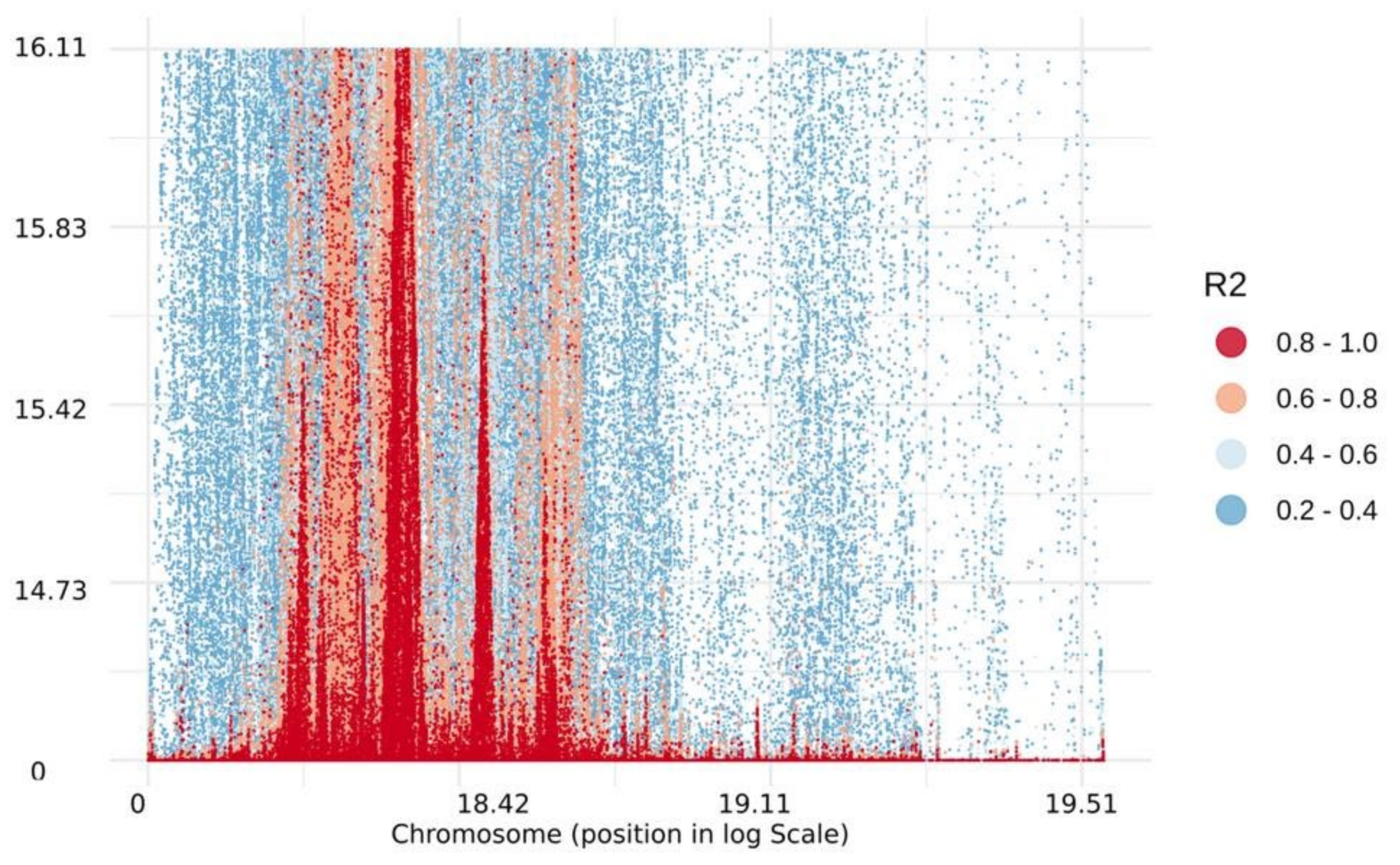
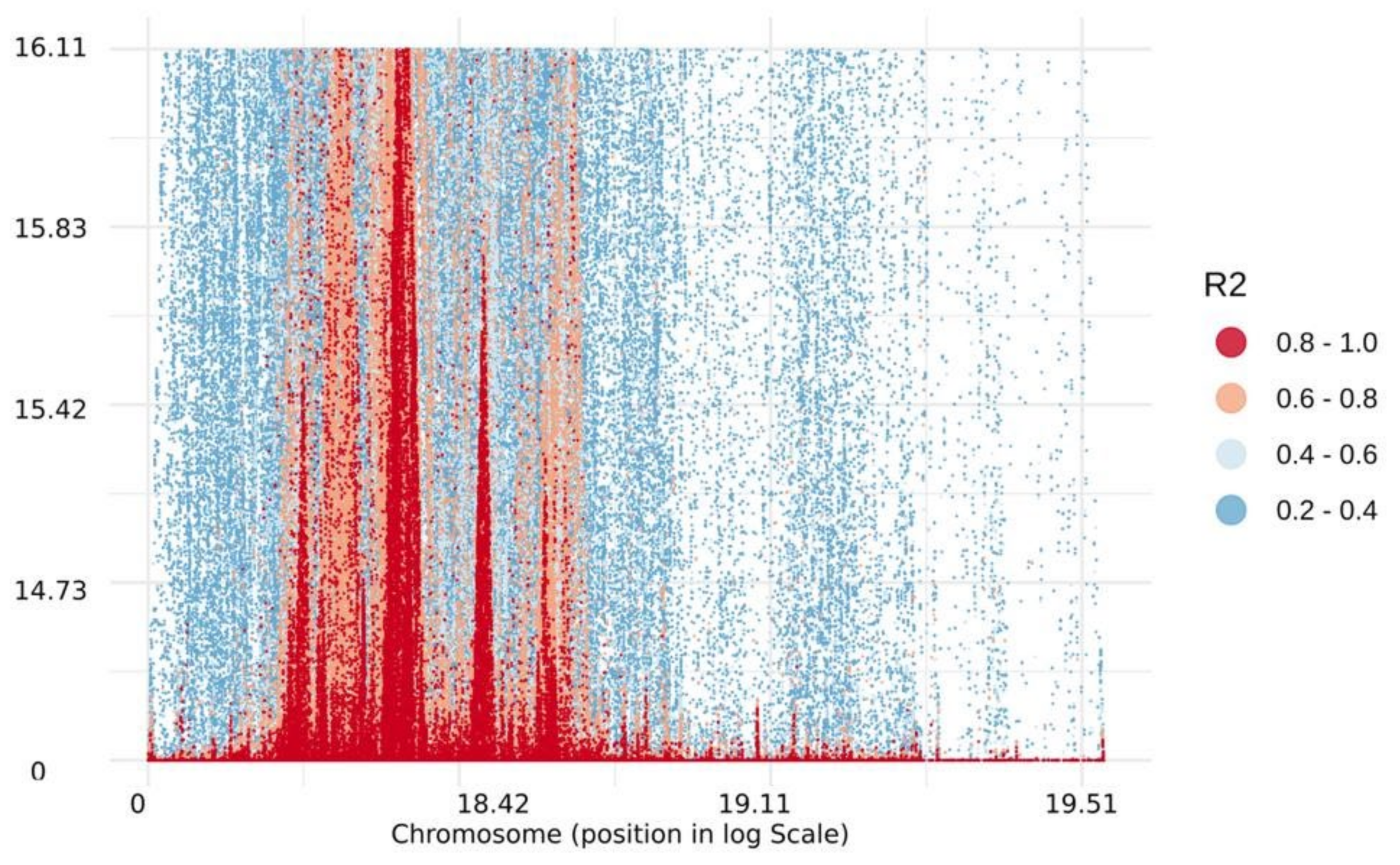
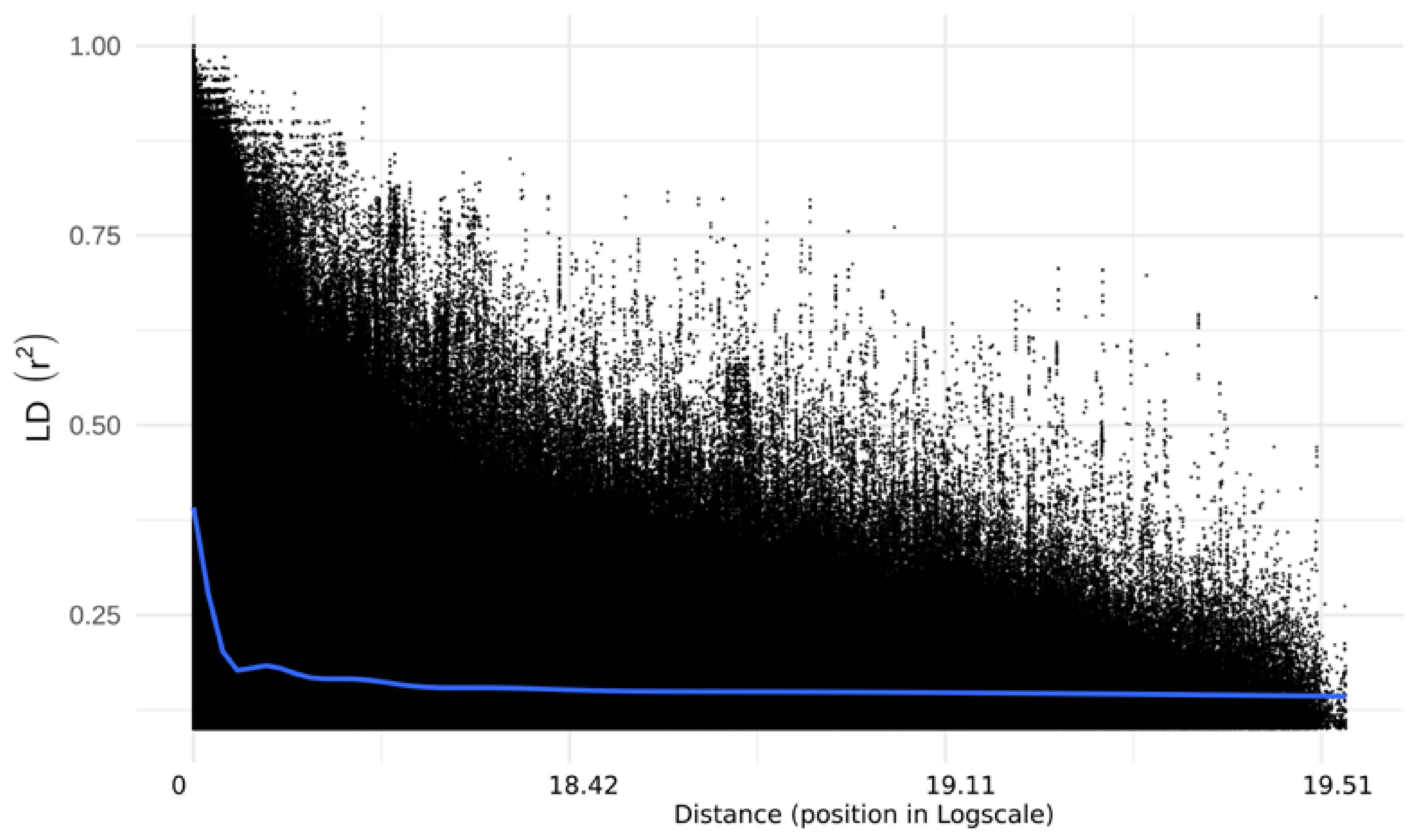
2.3. Linkage Disequilibrium and Haplotype Blocks Distribution
3. Discussion
3.1. Genetic Diversity and Population Structure
3.2. Linkage Disequilibrium and Distribution of Haplotype Blocks
4. Materials and Methods
4.1. Plant Material and DNA Isolation
4.2. Library Preparation and Genotyping
4.3. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harvey, M.; Pilgrim, S. The new competition for land: Food, energy, and climate change. Food Policy 2011, 36, 40–51. [Google Scholar] [CrossRef]
- Dass, S.; Kumar, A.; Jat, S.L.; Parihar, C.M.; Singh, A.K.; Chikkappa, G.K.; Jat, M.L. Maize holds potential for diversification and livelihood security. Indian J. Agron. 2012, 57, 32–37. [Google Scholar]
- Ranum, P.; Peña-Rosas, J.P.; Garcia-Casal, M.N. Global maize production, utilization, and consumption. Ann. N. Y. Acad. Sci. 2014, 1312, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Duo, H.; Hossain, F.; Muthusamy, V.; Zunjare, R.U.; Goswami, R.; Chand, G. Development of sub-tropically adapted diverse provitamin-A rich maize inbreds through marker assisted pedigree selection, their characterization and utilization in hybrid breeding. PLoS ONE 2021, 16, e0245497. [Google Scholar] [CrossRef]
- Murdia, L.K.; Wadhwani, R.; Wadhawan, N.; Bajpai, P.; Shekhawat, S. Maize utilization in India: An overview. Am. J. Food Nutr. 2016, 4, 169–176. [Google Scholar]
- Tewodros, M.; Mohammed, A. Heterotic response in major cereals and vegetable crops. Int. J. Plant Breed. Genet. 2016, 10, 69–78. [Google Scholar]
- Srivastava, K.R.; Bollam, S.; Pujarula, V.; Pusuluri, M.; Singh, R.B.; Potupureddi, G.; Gupta, R. Exploitation of heterosis in pearl millet: A review. Plants 2020, 9, 807. [Google Scholar] [CrossRef]
- Shu, G.; Cao, G.; Li, N.; Wang, A.; Wei, F.; Li, T.; Yi, L.; Xu, Y.; Wang, Y. Genetic variation and population structure in China summer maize germplasm. Sci. Rep. 2021, 11, 8012. [Google Scholar] [CrossRef]
- Ahmar, S.; Gill, R.A.; Jung, K.H.; Faheem, A.; Qasim, M.U.; Mubeen, M.; Zhou, W. Conventional and Molecular Techniques from Simple Breeding to Speed Breeding in Crop Plants: Recent Advances and Future Outlook. Int. J. Mol. Sci. 2020, 21, 2590. [Google Scholar] [CrossRef] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.X.; Liu, H.; Li, Z. Genotyping-by-sequencing (GBS), An ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.S.; Choi, S.C.; Jun, T.H.; Kim, C. Genotyping-by-sequencing: A promising tool for plant genetics research and breeding. Hortic. Environ. Biotechnol. 2017, 58, 425–431. [Google Scholar] [CrossRef]
- Ertiro, B.T.; Ogugo, V.; Worku, M.; Das, B.; Olsen, M.; Labuschagne, M.; Semagn, K. Comparison of Kompetitive Allele Specific PCR (KASP) and genotyping by sequencing (GBS) for quality control analysis in maize. BMC Genom. 2015, 16, 908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; San Vicente, F.; Huang, K.; Dhliwayo, T.; Costich, D.E.; Semagn, K.; Sudha, N.; Olsen, M.; Prasanna, B.M.; Zhang, X.; et al. Molecular characterization of CIMMYT maize inbred lines with genotyping-by-sequencing SNPs. Theor. Appl. Genet. 2016, 129, 753–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ertiro, B.T.; Semagn, K.; Das, B.; Olsen, M.; Labuschagne, M.; Worku, M.; Wegary, D.; Azmach, G.; Ogugo, V.; Keno, T.; et al. Genetic variation and population structure of maize inbred lines adapted to the mid-altitude sub-humid maize agro-ecology of Ethiopia using single nucleotide polymorphic (SNP) markers. BMC Genom. 2017, 18, 777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegary, D.; Teklewold, A.; Prasanna, B.M.; Ertiro, B.T.; Alachiotis, N.; Negera, D.; Awas, G.; Abakemal, D.; Ogugo, V.; Gowda, M.; et al. Molecular diversity and selective sweeps in maize inbred lines adapted to African highlands. Sci. Rep. 2019, 9, 13490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouesnard, B.; Negro, S.; Laffray, A.; Glaubitz, J.; Melchinger, A.; Revilla, P.; Moreno-Gonzalez, J.; Madur, D.; Combes, V.; Tollon-Cordet, C.; et al. Genotyping-by-sequencing highlights original diversity patterns within a European collection of 1191 maize flint lines, as compared to the maize USDA genebank. Theor. Appl. Genet. 2017, 130, 2165–2189. [Google Scholar] [CrossRef] [PubMed]
- Wasala, S.K.; Prasanna, B.M. Microsatellite marker-based diversity and population genetic analysis of selected lowland and mid-altitude maize landrace accessions of India. J. Plant Biochem. Biotechnol. 2013, 22, 392–400. [Google Scholar] [CrossRef]
- Sharma, L.; Prasanna, B.M.; Ramesh, B. Analysis of phenotypic and microsatellite-based diversity of maize landraces in India, especially from the North East Himalayan region. Genetica 2010, 138, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Mahar, K.S.; Agrawal, P.K.; Babu, B.K.; Gupta, H.S. Assessment of genetic diversity among the elite maize (Zea mays L.) genotypes adapted to North-Western Himalayan region of India using microsatellite markers. J. Plant Biochem. Biotechnol. 2009, 18, 217–220. [Google Scholar] [CrossRef]
- Sathua, S.K.; Shahi, J.P.; Mahato, A.; Gayatonde, V.; Kumar, P. Molecular diversity analysis of maize (Zea mays L.) inbreds using SSR markers. Electron. J. Plant Breed. 2018, 9, 1122–1129. [Google Scholar] [CrossRef]
- Singode, A.; Prasanna, B.M. Analysis of genetic diversity in the North Eastern Himalayan Maize landraces using microsatellite markers. J. Plant Biochem. Biotechnol. 2010, 19, 33–41. [Google Scholar] [CrossRef]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R.; Kumpatla, S. SNP markers and their impact on plant breeding. Int. J. Plant Genom. 2012, 2012, 728398. [Google Scholar] [CrossRef]
- Prasanna, B.M. Diversity in global maize germplasm: Characterization and utilization. J. Biosci. 2012, 37, 843–855. [Google Scholar] [CrossRef]
- Hall, D.; Zhao, W.; Wennström, U.; Andersson Gull, B.; Wang, X.-R. Parentage and relatedness reconstruction in Pinus sylvestris using genotyping-by-sequencing. Heredity 2020, 124, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Wang, W.; Gong, S.; Zuo, J.; Li, S.; Xu, S. High density linkage map construction and mapping of yield trait QTLs in maize (Zea mays) using the genotyping-by-sequencing (GBS) technology. Front. Plant Sci. 2017, 8, 706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafalski, J.A. Association genetics in crop improvement. Curr. Opin. Plant Biol. 2010, 13, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Slatkin, M. Linkage disequilibrium-understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 2008, 9, 477–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnable, P.S.; Ware, D.; Fulton, R.S. The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science 2012, 326, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Boakyewaa Adu, G.; Badu-Apraku, B.; Akromah, R.; Garcia-Oliveira, A.L.; Awuku, F.J.; Gedil, M. Genetic diversity and population structure of early-maturing tropical maize inbred lines using SNP markers. PLoS ONE 2019, 14, e0214810. [Google Scholar] [CrossRef] [Green Version]
- Dao, A.; Sanou, J.; Mitchell, S.E.; Gracen, V.; Danquah, E.Y. Genetic diversity among INERA maize inbred lines with single nucleotide polymorphism (SNP) markers and their relationship with CIMMYT, IITA, and temperate lines. BMC Genet. 2014, 15, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senior, M.L.; Murphy, J.P.; Goodman, M.M.; Stuber, C.W. Utility of SSRs for determining genetic similarities an relationships in maize using an agarose gel system. Crop Sci. 1998, 38, 1088–1098. [Google Scholar] [CrossRef]
- Cui, C.; Mei, H.; Liu, Y.; Zhang, H.; Zheng, Y. Genetic diversity, population structure, and linkage disequilibrium of an association-mapping panel revealed by genome-wide SNP markers in sesame. Front. Plant Sci. 2017, 8, 1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semagn, K.; Beyene, Y.; Makumbi, D.; Mugo, S.; Prasanna, B.M.; Magorokosho, C.; Atlin, G. Quality control genotyping for assessment of genetic identity and purity in diverse tropical maize inbred lines. Theor. Appl. Genet. 2012, 125, 1487–1501. [Google Scholar] [CrossRef]
- Xie, C.; Warburton, M.; Li, M.; Li, X.; Xiao, M.; Hao, Z.; Zhao, Q.; Zhang, S. Retracted Article: An analysis of population structure and linkage disequilibrium using multilocus data in 187 maize inbred lines. Mol. Breed. 2008, 21, 407–418. [Google Scholar] [CrossRef]
- Vigouroux, Y.; Glaubitz, J.C.; Matsuoka, Y.; Goodman, M.M.; Sánchez, G.J.; Doebley, J. Population structure and genetic diversity of New World maize races assessed by DNA microsatellites. Am. J. Bot. 2008, 95, 1240–1253. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Shah, T.; Warburton, M.L.; Buckler, E.S.; McMullen, M.D.; Crouch, J. Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS ONE 2009, 4, e8451. [Google Scholar] [CrossRef] [Green Version]
- Remington, D.L.; Thornsberry, J.M.; Matsuoka, Y.; Wilson, L.M.; Whitt, S.R.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 2001, 98, 11479–11484. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Lv, X.; Weng, J.; Zhu, H.; Liu, C.; Hao, Z.; Zhou, Y.; Zhang, D.; Li, M.; Ci, X. Genetic characterization and linkage disequilibrium mapping of resistance to gray leaf spot in maize (Zea mays L.). Crop J. 2014, 2, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Rakshit, S.; Rakshit, A.; Matsumura, H.; Takahashi, Y.; Hasegawa, Y.; Ito, A.; Ishii, T.; Miyashita, N.T.; Terauchi, R. Large-scale DNA polymorphism study of Oryza sativa and O. rufipogon reveals the origin and divergence of Asian rice. Theor. Appl. Genet. 2007, 114, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebbert, M.T.W.; Wadsworth, M.E.; Staley, L.A. Evaluating the necessity of PCR duplicate removal from next-generation sequencing data and a comparison of approaches. BMC Bioinform. 2016, 17, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Wu, X.; Li, Y.; Shi, Y.; Song, Y.; Wang, T.; Huang, Y.; Li, Y. Fine genetic characterization of elite maize germplasm using high-throughput SNP genotyping. Theor. Appl. Genet. 2014, 127, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Leng, Y.; Lv, C.; Li, L.; Xiang, Y.; Xia, C.; Wei, R.; Rong, T.; Lan, H. Heterotic grouping based on genetic variation and population structure of maize inbred lines from current breeding program in Sichuan province, Southwest China using genotyping by sequencing (GBS). Mol. Breed. 2019, 39, 38. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Weir, B.S. Genetic Data Analysis II; Sinauer Associates, Inc. Publishers: Sunderland, MA, USA, 1996. [Google Scholar]
- Otyama, P.I. Evaluation of linkage disequilibrium, population structure, and genetic diversity in the US peanut mini core collection. BMC Genom. 2019, 20, 481. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 633–2635. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome (Chr.) | Physical Map Length (Mb) | Number of SNPs at MAF 0.05 | Number of Loci | SNPs at MAF (≥0.1) |
|---|---|---|---|---|
| Chr-1 | 307.04 | 8976 | 2906 | 6946 |
| Chr-2 | 244.44 | 7173 | 2299 | 5377 |
| Chr-3 | 235.67 | 7405 | 2353 | 5757 |
| Chr-4 | 246.99 | 8293 | 2676 | 6270 |
| Chr-5 | 223.9 | 6823 | 2270 | 5142 |
| Chr-6 | 174.03 | 4624 | 1511 | 3523 |
| Chr-7 | 182.38 | 6053 | 1983 | 4456 |
| Chr-8 | 181.12 | 5345 | 1733 | 4134 |
| Chr-9 | 110.77 | 3368 | 1080 | 2618 |
| Chr-10 | 150.98 | 2167 | 1274 | 1325 |
| Total | 2057.32 | 60,227 | 20,085 | 45,548 |
| Parameter | Value | Type of Mutation | SNP Mutation | Number of SNPs | Total SNPs per Type of Mutation |
|---|---|---|---|---|---|
| Number of SNPs | 60,227 | Transitions | A/G | 21,775 | 43,701 |
| Ts/Tv ratio | 2.64 | C/T | 21,926 | ||
| Mean Nucleotide Diversity | 0.28 | Transversions | A/T | 4435 | 16,526 |
| A/C | 4505 | ||||
| Frequency of polymorphic sites per bp | 0.02 | C/G | 2996 | ||
| G/T | 4590 | ||||
| Proportion of heterogenous SNPs | 0.17 | Total | 60,227 | 60,227 |
| Chr. | Chr. Length (Mb) | Hap. Freq (%) | a Nohb | Total Block Length (kb) | b Nohb | c Sum (>100 kb) | d TSNP | e Max (kb) | f Max SNPs | g Min SNPs |
|---|---|---|---|---|---|---|---|---|---|---|
| Chr1 | 307.04 | 30.86 | 1556 | 9220 | 33 | 4894.75 | 5614 (334) | 198.16 | 28 | 2 |
| Chr2 | 244.44 | 30.01 | 1182 | 8740 | 37 | 5385.36 | 4378 (342) | 199.69 | 27 | 2 |
| Chr3 | 235.67 | 30.40 | 1282 | 11,470 | 45 | 6800.93 | 4784 (384) | 199.28 | 26 | 2 |
| Chr4 | 246.99 | 30.87 | 1432 | 9040 | 33 | 4843.49 | 5184 (272) | 193.46 | 19 | 2 |
| Chr5 | 223.90 | 31.08 | 1137 | 7160 | 29 | 3978.95 | 4198 (276) | 196.90 | 22 | 2 |
| Chr6 | 174.03 | 30.97 | 799 | 3410 | 11 | 1577.28 | 2767 (133) | 193.12 | 25 | 2 |
| Chr7 | 182.38 | 30.08 | 1065 | 9620 | 43 | 6333.56 | 3888 (387) | 197.22 | 30 | 2 |
| Chr8 | 181.12 | 30.89 | 876 | 6990 | 30 | 4317.02 | 3420 (346) | 199.99 | 23 | 2 |
| Chr9 | 110.77 | 30.35 | 586 | 3450 | 10 | 1559.55 | 2122 (96) | 198.88 | 18 | 2 |
| Chr10 | 150.98 | 36.35 | 285 | 880 | 3 | 473.303 | 744 (13) | 180.03 | 8 | 2 |
| Mean | 205.73 | 31.19 | 1020 | 6998 | 27 | 4016.42 | 3710 | 195.67 | 23 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, B.; Rakshit, S.; Kumar, S.; Singh, B.K.; Lahkar, C.; Jha, A.K.; Kumar, K.; Kumar, P.; Choudhary, M.; Singh, S.B.; et al. Genetic Diversity, Population Structure and Linkage Disequilibrium Analyses in Tropical Maize Using Genotyping by Sequencing. Plants 2022, 11, 799. https://doi.org/10.3390/plants11060799
Kumar B, Rakshit S, Kumar S, Singh BK, Lahkar C, Jha AK, Kumar K, Kumar P, Choudhary M, Singh SB, et al. Genetic Diversity, Population Structure and Linkage Disequilibrium Analyses in Tropical Maize Using Genotyping by Sequencing. Plants. 2022; 11(6):799. https://doi.org/10.3390/plants11060799
Chicago/Turabian StyleKumar, Bhupender, Sujay Rakshit, Sonu Kumar, Brijesh Kumar Singh, Chayanika Lahkar, Abhishek Kumar Jha, Krishan Kumar, Pardeep Kumar, Mukesh Choudhary, Shyam Bir Singh, and et al. 2022. "Genetic Diversity, Population Structure and Linkage Disequilibrium Analyses in Tropical Maize Using Genotyping by Sequencing" Plants 11, no. 6: 799. https://doi.org/10.3390/plants11060799
APA StyleKumar, B., Rakshit, S., Kumar, S., Singh, B. K., Lahkar, C., Jha, A. K., Kumar, K., Kumar, P., Choudhary, M., Singh, S. B., Amalraj, J. J., Prakash, B., Khulbe, R., Kamboj, M. C., Chirravuri, N. N., & Hossain, F. (2022). Genetic Diversity, Population Structure and Linkage Disequilibrium Analyses in Tropical Maize Using Genotyping by Sequencing. Plants, 11(6), 799. https://doi.org/10.3390/plants11060799