
Genome-Wide Analysis and Characterization of the Proline-Rich Extensin-like Receptor Kinases (PERKs) Gene Family Reveals Their Role in Different Developmental Stages and Stress Conditions in Wheat (Triticum aestivum L.)

 , ,
, ,  ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Identification of TaPERK in Wheat
2.2. Chromosomal Distribution, Gene Duplication, and Synteny Analysis
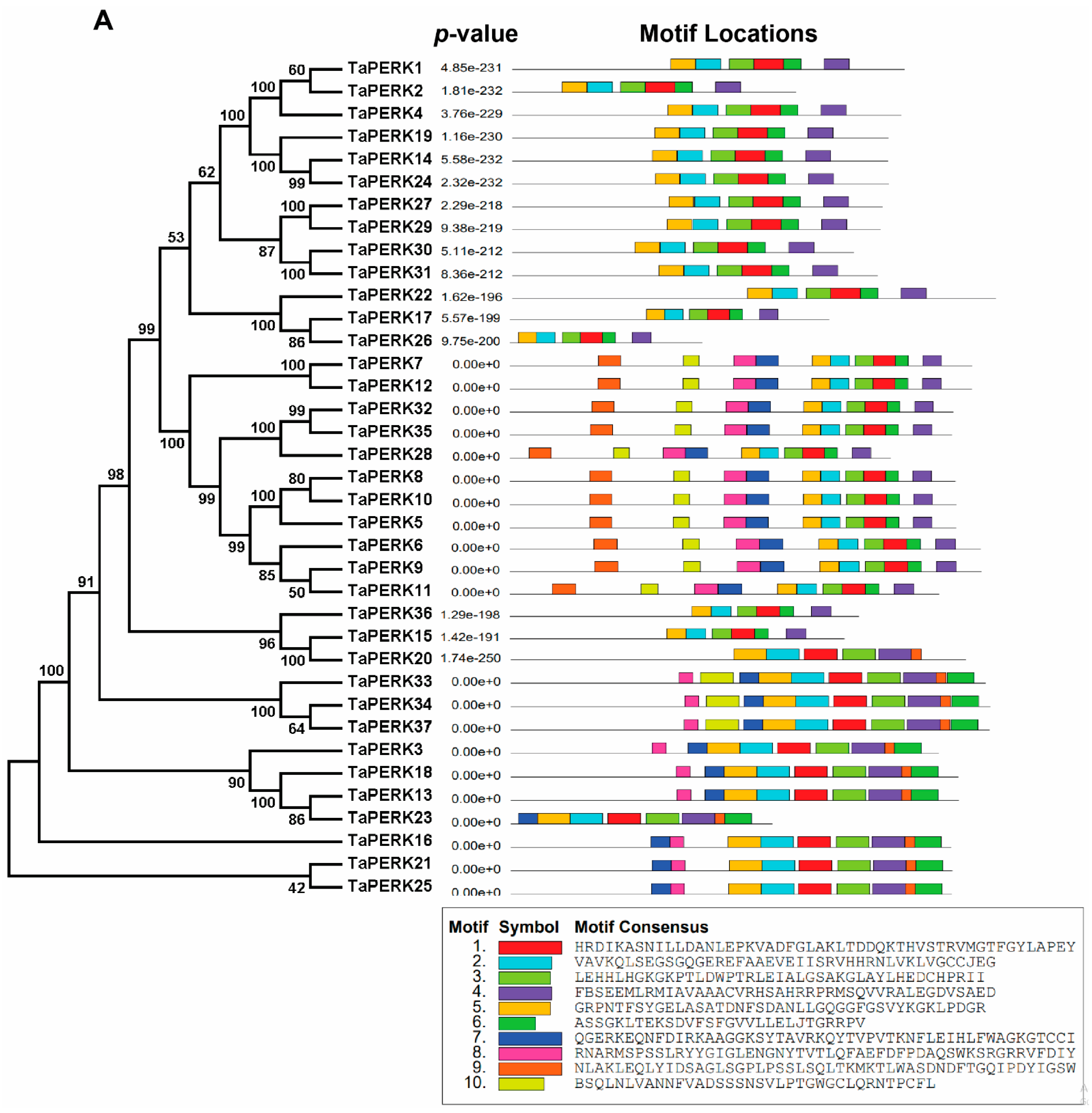
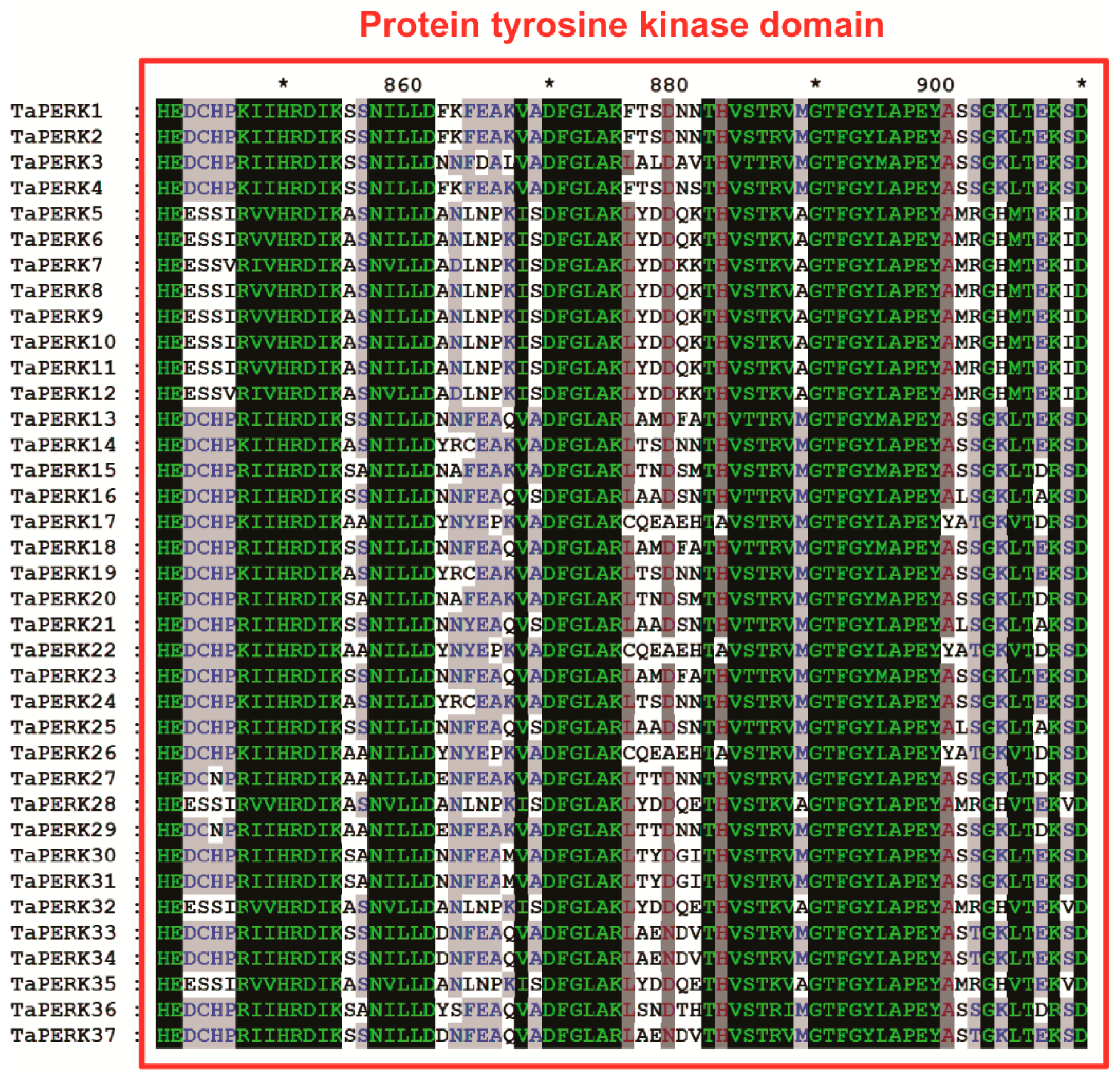
2.3. Exon/Intron Structure and Motif analysis of TaPERK Genes
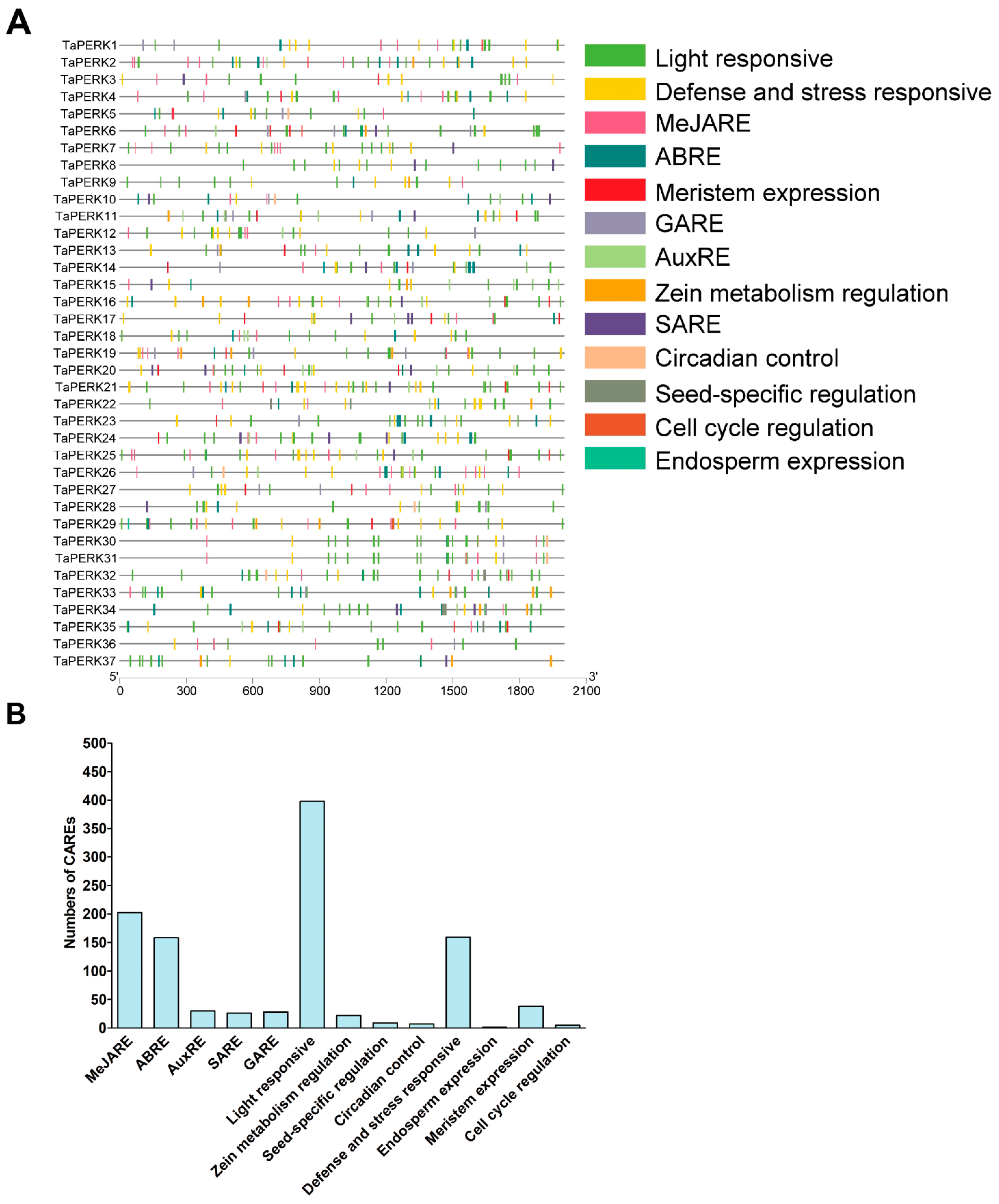
2.4. Cis-Acting Regulatory Elements (CAREs) Analysis of TaPERK Genes
2.5. Gene Ontology (GO) Enrichment of TaPERK Genes
2.6. Expression Profiling of TaPERK Genes in Various Developmental Stages and under Diverse Stress Conditions
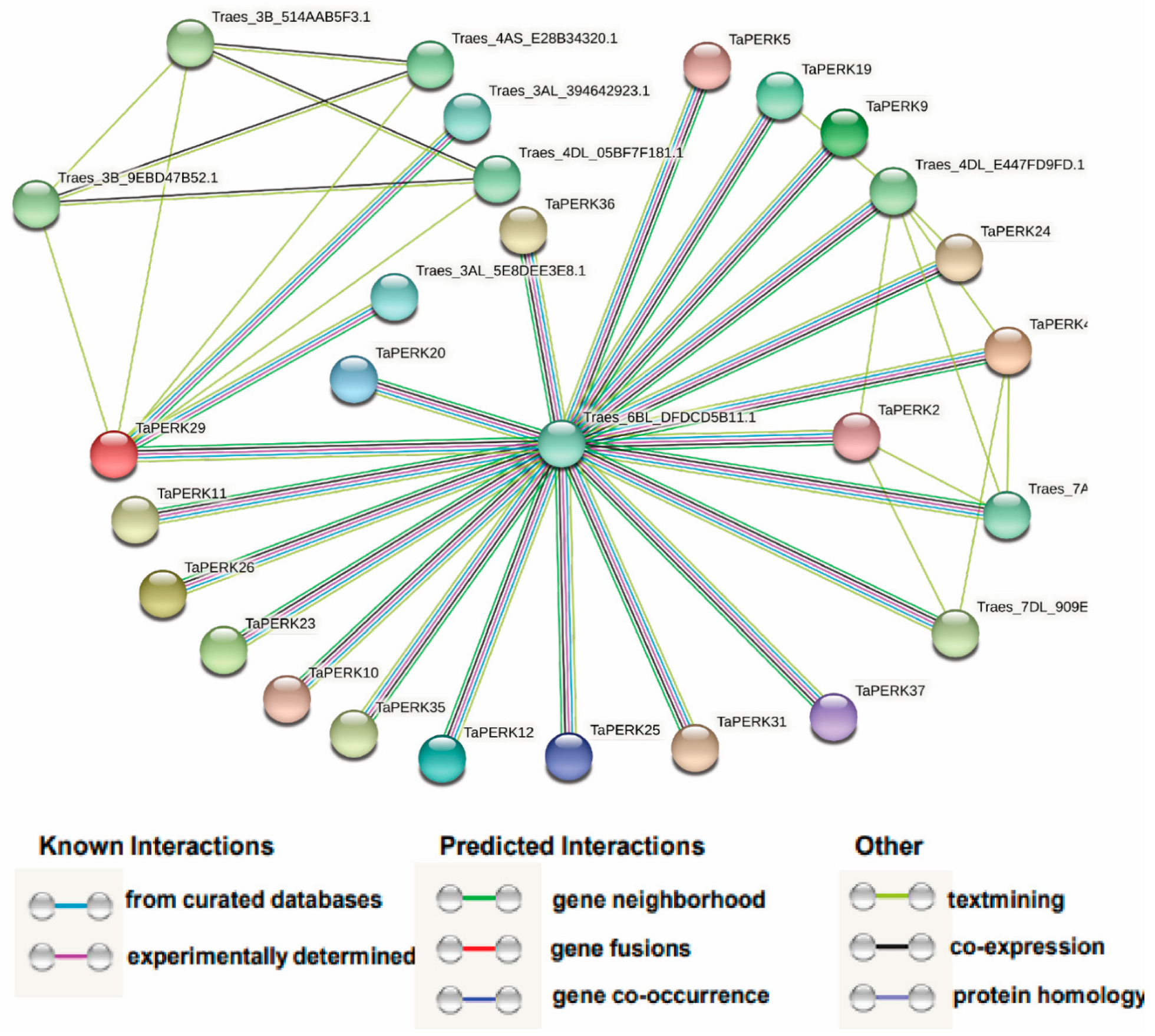
2.7. Protein–Protein Network Analysis of the TaPERK Family Genes
3. Discussion
4. Materials and Methods
4.1. Identification of PERK Genes in Wheat
4.2. Genomic Localization, Gene Duplication, and Synteny Analysis
4.3. Biophysical Characteristics, Subcellular Localization, and 3D Structure
4.4. Exon/intron Structure, Protein Motif, and Gene Ontology Analysis
4.5. Promoter Cis-Acting Regulatory Elements (CAREs) and Protein Interaction Network Analysis
4.6. Expression Analysis of TaPERK Genes
4.7. Plant Growth Conditions, Stress Treatment, and RT-qPCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Day, E.K.; Sosale, N.G.; Lazzara, M.J. Cell signaling regulation by protein phosphorylation: A multivariate, heterogeneous, and context-dependent process. Curr. Opin. Biotechnol. 2016, 40, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, U.; Lau, K.; Hothorn, M. The structural basis of ligand perception and signal activation by receptor kinases. Annu. Rev. Plant Biol. 2017, 68, 109–137. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.-S.; Kesawat, M.S.; Kumar, M.; Kim, S.-Y.; Mani, V.; Subramanian, P.; Park, S.; Lee, C.-M.; Kim, S.-R.; Hahn, B.-S. Lack of the α1, 3-fucosyltransferase gene (OsFucT) affects anther development and pollen viability in rice. Int. J. Mol. Sci. 2018, 19, 1225. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.K.; Quinn, A.M.; Hunter, T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988, 241, 42–52. [Google Scholar] [CrossRef]
- DeFalco, T.A.; Anne, P.; James, S.R.; Willoughby, A.; Johanndrees, O.; Genolet, Y.; Pullen, A.-M.; Zipfel, C.; Hardtke, C.S.; Nimchuk, Z.L. A conserved regulatory module regulates receptor kinase signaling in immunity and development. bioRxiv 2021. [Google Scholar] [CrossRef]
- Nakhamchik, A.; Zhao, Z.; Provart, N.J.; Shiu, S.-H.; Keatley, S.K.; Cameron, R.K.; Goring, D.R. A comprehensive expression analysis of the Arabidopsis proline-rich extensin-like receptor kinase gene family using bioinformatic and experimental approaches. Plant Cell Physiol. 2004, 45, 1875–1881. [Google Scholar] [CrossRef]
- Qanmber, G.; Liu, J.; Yu, D.; Liu, Z.; Lu, L.; Mo, H.; Ma, S.; Wang, Z.; Yang, Z. Genome-wide identification and characterization of the PERK gene family in Gossypium hirsutum reveals gene duplication and functional divergence. Int. J. Mol. Sci. 2019, 20, 1750. [Google Scholar] [CrossRef]
- Diévart, A.; Clark, S.E. LRR-containing receptors regulating plant development and defense. Development 2004, 131, 251–261. [Google Scholar] [CrossRef]
- Shiu, S.-H.; Bleecker, A.B. Receptor-like kinases from Arabidopsis form a monophyletic gene family related to animal receptor kinases. Proc. Natl. Acad. Sci. USA 2001, 98, 10763–10768. [Google Scholar] [CrossRef]
- Morris, E.R.; Walker, J.C. Receptor-like protein kinases: The keys to response. Curr. Opin. Plant Biol. 2003, 6, 339–342. [Google Scholar] [CrossRef]
- Shiu, S.-H.; Karlowski, W.M.; Pan, R.; Tzeng, Y.-H.; Mayer, K.F.; Li, W.-H. Comparative analysis of the receptor-like kinase family in Arabidopsis and rice. Plant Cell 2004, 16, 1220–1234. [Google Scholar] [CrossRef]
- Silva, N.F.; Goring, D.R. The proline-rich, extensin-like receptor kinase-1 (PERK1) gene is rapidly induced by wounding. Plant Mol. Biol. 2002, 50, 667–685. [Google Scholar] [CrossRef]
- Haffani, Y.Z.; Silva, N.F.; Goring, D.R. Receptor kinase signalling in plants. Can. J. Bot. 2004, 82, 1–15. [Google Scholar] [CrossRef][Green Version]
- Bai, L.; Zhang, G.; Zhou, Y.; Zhang, Z.; Wang, W.; Du, Y.; Wu, Z.; Song, C.P. Plasma membrane-associated proline-rich extensin-like receptor kinase 4, a novel regulator of Ca2+ signalling, is required for abscisic acid responses in Arabidopsis thaliana. Plant J. 2009, 60, 314–327. [Google Scholar] [CrossRef]
- Chen, G.; Wang, J.; Wang, H.; Wang, C.; Tang, X.; Li, J.; Zhang, L.; Song, J.; Hou, J.; Yuan, L. Genome-wide analysis of proline-rich extension-like receptor protein kinase (PERK) in Brassica rapa and its association with the pollen development. BMC Genom. 2020, 21, 401. [Google Scholar] [CrossRef]
- Borassi, C.; Sede, A.; Mecchia, M.A.; Mangano, S.; Marzol, E.; Denita-Juarez, S.P.; Salter, J.D.S.; Velasquez, S.M.; Muschietti, J.P.; Estevez, J. Proline-rich Extensin-like Receptor Kinases PERK5 and PERK12 are involved in Pollen Tube Growth. bioRxiv 2021. [Google Scholar] [CrossRef]
- Shiu, S.-H.; Bleecker, A.B. Expansion of the receptor-like kinase/Pelle gene family and receptor-like proteins in Arabidopsis. Plant Physiol. 2003, 132, 530–543. [Google Scholar] [CrossRef]
- Li, J.; Chory, J. A putative leucine-rich repeat receptor kinase involved in brassinosteroid signal transduction. Cell 1997, 90, 929–938. [Google Scholar] [CrossRef]
- Li, J.; Wen, J.; Lease, K.A.; Doke, J.T.; Tax, F.E.; Walker, J.C. BAK1, an Arabidopsis LRR receptor-like protein kinase, interacts with BRI1 and modulates brassinosteroid signaling. Cell 2002, 110, 213–222. [Google Scholar] [CrossRef]
- Nam, K.H.; Li, J. BRI1/BAK1, a receptor kinase pair mediating brassinosteroid signaling. Cell 2002, 110, 203–212. [Google Scholar] [CrossRef]
- Haffani, Y.; Silva-Gagliardi, N.; Sewter, S.; Aldea, M.G.; Zhao, Z.; Nakhamchik, A.; Cameron, R.; Goring, D. Altered expression of PERK receptor kinases in Arabidopsis leads to changes in growth and floral organ formation. Plant Signal. Behav. 2006, 1, 251–260. [Google Scholar] [CrossRef]
- Won, S.-K.; Lee, Y.-J.; Lee, H.-Y.; Heo, Y.-K.; Cho, M.; Cho, H.-T. Cis-element-and transcriptome-based screening of root hair-specific genes and their functional characterization in Arabidopsis. Plant Physiol. 2009, 150, 1459–1473. [Google Scholar] [CrossRef]
- Humphrey, T.V.; Haasen, K.E.; Aldea-Brydges, M.G.; Sun, H.; Zayed, Y.; Indriolo, E.; Goring, D.R. PERK–KIPK–KCBP signalling negatively regulates root growth in Arabidopsis thaliana. J. Exp. Bot. 2015, 66, 71–83. [Google Scholar] [CrossRef]
- Hwang, I.; Kim, S.Y.; Kim, C.S.; Park, Y.; Tripathi, G.R.; Kim, S.-K.; Cheong, H. Over-expression of the IGI1 leading to altered shoot-branching development related to MAX pathway in Arabidopsis. Plant Mol. Biol. 2010, 73, 629–641. [Google Scholar] [CrossRef][Green Version]
- Borassi, C.; Sede, A.R.; Mecchia, M.A.; Salgado Salter, J.D.; Marzol, E.; Muschietti, J.P.; Estevez, J.M. An update on cell surface proteins containing extensin-motifs. J. Exp. Bot. 2016, 67, 477–487. [Google Scholar] [CrossRef]
- Mansfield, T.; Hetherington, A.; Atkinson, C. Some current aspects of stomatal physiology. Annu. Rev. Plant Biol. 1990, 41, 55–75. [Google Scholar] [CrossRef]
- Webb, A.A.; McAinsh, M.R.; Taylor, J.E.; Hetherington, A.M. Calcium ions as intracellular second messengers in higher plants. Adv. Bot. Res. 1996, 22, 45–96. [Google Scholar] [CrossRef]
- Gong, M.; Chen, S.-N.; Song, Y.-Q.; Li, Z.-G. Effect of calcium and calmodulin on intrinsic heat tolerance in relation to antioxidant systems in maize seedlings. Funct. Plant Biol. 1997, 24, 371–379. [Google Scholar] [CrossRef]
- Hwang, Y.; Lee, H.; Lee, Y.S.; Cho, H.T. Cell wall-associated ROOT HAIR SPECIFIC 10, a proline-rich receptor-like kinase, is a negative modulator of Arabidopsis root hair growth. J. Exp. Bot. 2016, 67, 2007–2022. [Google Scholar] [CrossRef]
- Samuel, M.A.; Ellis, B.E. Double jeopardy: Both overexpression and suppression of a redox-activated plant mitogen-activated protein kinase render tobacco plants ozone sensitive. Plant Cell 2002, 14, 2059–2069. [Google Scholar] [CrossRef]
- Xing, Y.; Cao, Q.Q.; Zhang, Q.; Qin, L.; Jia, W.S.; Zhang, J.H. MKK5 Regulates High Light-Induced Gene Expression of Cu/Zn Superoxide Dismutase 1 and 2 in Arabidopsis. Plant Cell Physiol. 2013, 54, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Florentino, L.H.; Santos, A.A.; Fontenelle, M.R.; Pinheiro, G.L.; Zerbini, F.M.; Baracat-Pereira, M.C.; Fontes, E.P. A PERK-like receptor kinase interacts with the geminivirus nuclear shuttle protein and potentiates viral infection. J. Virol. 2006, 80, 6648–6656. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.M.; Kumar, D.B.; Kumar, D.S. Manorama. Assessment of genetic diversity among Indian Sesame (Sesamum indicum L.) accessions using RAPD, ISSR and SSR markers. Res. J. Biotechnol. 2015, 10, 35–47. [Google Scholar]
- Kumar, M.; Kesawat, M.S.; Ali, A.; Lee, S.-C.; Gill, S.S.; Kim, H.U. Integration of abscisic acid signaling with other signaling pathways in plant stress responses and development. Plants 2019, 8, 592. [Google Scholar] [CrossRef] [PubMed]
- Kesawat, M.S.; Shivaraj, S.; Kim, D.K.; Kumar, M.; Hahn, B.S.; Deshmukh, R. Metalloids and Their Role in the Biological System. Met. Plants Adv. Future Prospect. 2020, 1–17. [Google Scholar] [CrossRef]
- The International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar] [CrossRef]
- Gill, B.S.; Appels, R.; Botha-Oberholster, A.-M.; Buell, C.R.; Bennetzen, J.L.; Chalhoub, B.; Chumley, F.; Dvorák, J.; Iwanaga, M.; Keller, B. A workshop report on wheat genome sequencing: International Genome Research on Wheat Consortium. Genetics 2004, 168, 1087–1096. [Google Scholar] [CrossRef]
- Afzal, F.; Chaudhari, S.K.; Gul, A.; Farooq, A.; Ali, H.; Nisar, S.; Sarfraz, B.; Shehzadi, K.J.; Mujeeb-Kazi, A. Bread wheat (Triticum aestivum L.) under biotic and abiotic stresses: An overview. Crop Prod. Glob. Environ. Issues 2015, 293–317. [Google Scholar] [CrossRef]
- Kumar, P.; Yadava, R.; Gollen, B.; Kumar, S.; Verma, R.K.; Yadav, S. Nutritional contents and medicinal properties of wheat: A review. Life Sci. Med. Res. 2011, 22, 1–10. [Google Scholar]
- Kumar, M.; Kherawat, B.S.; Dey, P.; Saha, D.; Singh, A.; Bhatia, S.K.; Ghodake, G.S.; Kadam, A.A.; Kim, H.-U.; Chung, S.-M. Genome-Wide Identification and Characterization of PIN-FORMED (PIN) Gene Family Reveals Role in Developmental and Various Stress Conditions in Triticum aestivum L. Int. J. Mol. Sci. 2021, 22, 7396. [Google Scholar] [CrossRef]
- Kesawat, M.S.; Das, B.K.; Bhaganagare, G.R. Genome-wide identification, evolutionary and expression analyses of putative Fe–S biogenesis genes in rice (Oryza sativa). Genome 2012, 55, 571–583. [Google Scholar] [CrossRef]
- Kesawat, M.S.; Kherawat, B.S.; Singh, A.; Dey, P.; Kabi, M.; Debnath, D.; Saha, D.; Khandual, A.; Rout, S.; Ali, A. Genome-wide identification and characterization of the brassinazole-resistant (BZR) gene family and its expression in the various developmental stage and stress conditions in wheat (Triticum aestivum L.). Int. J. Mol. Sci. 2021, 22, 8743. [Google Scholar] [CrossRef]
- Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; Poland, J. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361. [Google Scholar] [CrossRef]
- Lawton-Rauh, A. Evolutionary dynamics of duplicated genes in plants. Mol. Phylogenet. Evol. 2003, 29, 396–409. [Google Scholar] [CrossRef]
- Hou, Y.; Gupta, N.; Schoenlein, P.; Wong, E.; Martindale, R.; Ganapathy, V.; Browning, D. An anti-tumor role for cGMP-dependent protein kinase. Cancer Lett. 2006, 240, 60–68. [Google Scholar] [CrossRef]
- Gross, I.; Durner, J. In search of enzymes with a role in 3′, 5′-cyclic guanosine monophosphate metabolism in plants. Front. Plant Sci. 2016, 7, 576. [Google Scholar] [CrossRef]
- Shen, Q.; Zhan, X.; Yang, P.; Li, J.; Chen, J.; Tang, B.; Wang, X.; Hong, Y. Dual activities of plant cGMP-dependent protein kinase and its roles in gibberellin signaling and salt stress. Plant Cell 2019, 31, 3073–3091. [Google Scholar] [CrossRef]
- Wheeler, J.I.; Wong, A.; Marondedze, C.; Groen, A.J.; Kwezi, L.; Freihat, L.; Vyas, J.; Raji, M.A.; Irving, H.R.; Gehring, C. The brassinosteroid receptor BRI 1 can generate cGMP enabling cGMP-dependent downstream signaling. Plant J. 2017, 91, 590–600. [Google Scholar] [CrossRef]
- Isner, J.-C.; Maathuis, F.J. cGMP signalling in plants: From enigma to main stream. Funct. Plant Biol. 2016, 45, 93–101. [Google Scholar] [CrossRef]
- Lespinet, O.; Wolf, Y.I.; Koonin, E.V.; Aravind, L. The role of lineage-specific gene family expansion in the evolution of eukaryotes. Genome Res. 2002, 12, 1048–1059. [Google Scholar] [CrossRef]
- Jordan, I.K.; Makarova, K.S.; Spouge, J.L.; Wolf, Y.I.; Koonin, E.V. Lineage-specific gene expansions in bacterial and archaeal genomes. Genome Res. 2001, 11, 555–565. [Google Scholar] [CrossRef]
- Ramsey, J.; Schemske, D.W. Pathways, mechanisms, and rates of polyploid formation in flowering plants. Annu. Rev. Ecol. Syst. 1998, 29, 467–501. [Google Scholar] [CrossRef]
- Moore, R.C.; Purugganan, M.D. The early stages of duplicate gene evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 15682–15687. [Google Scholar] [CrossRef]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef]
- Baumberger, N.; Doesseger, B.; Guyot, R.; Diet, A.; Parsons, R.L.; Clark, M.A.; Simmons, M.; Bedinger, P.; Goff, S.A.; Ringli, C. Whole-genome comparison of leucine-rich repeat extensins in Arabidopsis and rice. A conserved family of cell wall proteins form a vegetative and a reproductive clade. Plant Physiol. 2003, 131, 1313–1326. [Google Scholar] [CrossRef]
- Wang, D.; Guo, Y.; Wu, C.; Yang, G.; Li, Y.; Zheng, C. Genome-wide analysis of CCCH zinc finger family in Arabidopsis and rice. BMC Genom. 2008, 9, 44. [Google Scholar] [CrossRef]
- Yang, Z.; Gong, Q.; Qin, W.; Yang, Z.; Cheng, Y.; Lu, L.; Ge, X.; Zhang, C.; Wu, Z.; Li, F. Genome-wide analysis of WOX genes in upland cotton and their expression pattern under different stresses. BMC Plant Biol. 2017, 17, 113. [Google Scholar] [CrossRef]
- Yang, Z.; Gong, Q.; Wang, L.; Jin, Y.; Xi, J.; Li, Z.; Qin, W.; Yang, Z.; Lu, L.; Chen, Q. Genome-wide study of YABBY genes in upland cotton and their expression patterns under different stresses. Front. Genet. 2018, 9, 33. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.; Yang, Z.E.; Chen, E.Y.; Zhang, C.J.; Zhang, X.Y.; Li, F.G. Genome-wide analysis of GRAS transcription factor gene family in Gossypium hirsutum L. BMC Genom. 2018, 19, 348. [Google Scholar] [CrossRef]
- Yin, G.; Xu, H.; Xiao, S.; Qin, Y.; Li, Y.; Yan, Y.; Hu, Y. The large soybean (Glycine max) WRKY TF family expanded by segmental duplication events and subsequent divergent selection among subgroups. BMC Plant Biol. 2013, 13, 148. [Google Scholar] [CrossRef]
- Ren, Z.; Yu, D.; Yang, Z.; Li, C.; Qanmber, G.; Li, Y.; Li, J.; Liu, Z.; Lu, L.; Wang, L. Genome-wide identification of the MIKC-type MADS-box gene family in Gossypium hirsutum L. unravels their roles in flowering. Front. Plant Sci. 2017, 8, 384. [Google Scholar] [CrossRef] [PubMed]
- Dossa, K.; Diouf, D.; Cissé, N. Genome-wide investigation of Hsf genes in sesame reveals their segmental duplication expansion and their active role in drought stress response. Front. Plant Sci. 2016, 7, 1522. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.W.; Gilbert, W. The evolution of spliceosomal introns: Patterns, puzzles and progress. Nat. Rev. Genet. 2006, 7, 211–221. [Google Scholar] [PubMed]
- Roy, S.W.; Penny, D. Patterns of intron loss and gain in plants: Intron loss–dominated evolution and genome-wide comparison of O. sativa and A. thaliana. Mol. Biol. Evol. 2007, 24, 171–181. [Google Scholar] [CrossRef]
- Serrano, M.; Parra, S.; Alcaraz, L.D.; Guzmán, P. The ATL gene family from Arabidopsis thaliana and Oryza sativa comprises a large number of putative ubiquitin ligases of the RING-H2 type. J. Mol. Evol. 2006, 62, 434–445. [Google Scholar] [CrossRef]
- Dan, Y.; Niu, Y.; Wang, C.; Yan, M.; Liao, W. Genome-wide identification and expression analysis of the trehalose-6-phosphate synthase (TPS) gene family in cucumber (Cucumis sativus L.). PeerJ 2021, 9, e11398. [Google Scholar] [CrossRef]
- Lecharny, A.; Boudet, N.; Gy, I.; Aubourg, S.; Kreis, M. Introns in, introns out in plant gene families: A genomic approach of the dynamics of gene structure. J. Struct. Funct. Genom. 2003, 3, 111–116. [Google Scholar] [CrossRef]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and validation of promoters and cis-acting regulatory elements. Plant Sci. 2014, 217, 109–119. [Google Scholar] [CrossRef]
- Roy, A.L.; Sen, R.; Roeder, R.G. Enhancer–promoter communication and transcriptional regulation of Igh. Trends Immunol. 2011, 32, 532–539. [Google Scholar] [CrossRef]
- Roy, A.L.; Singer, D.S. Core promoters in transcription: Old problem, new insights. Trends Biochem. Sci. 2015, 40, 165–171. [Google Scholar] [CrossRef]
- Zhang, Y.; Wong, C.-H.; Birnbaum, R.Y.; Li, G.; Favaro, R.; Ngan, C.Y.; Lim, J.; Tai, E.; Poh, H.M.; Wong, E. Chromatin connectivity maps reveal dynamic promoter–enhancer long-range associations. Nature 2013, 504, 306–310. [Google Scholar] [CrossRef]
- Cheng, Y.; Tang, Q.; Li, Y.; Zhang, Y.; Zhao, C.; Yan, J.; You, H. Folding/unfolding kinetics of G-quadruplexes upstream of the P1 promoter of the human BCL-2 oncogene. J. Biol. Chem. 2019, 294, 5890–5895. [Google Scholar] [CrossRef]
- Fankhauser, C.; Chory, J. Light control of plant development. Annu. Rev. Cell Dev. Biol. 1997, 13, 203–229. [Google Scholar] [CrossRef]
- Dievart, A.; Gottin, C.; Périn, C.; Ranwez, V.; Chantret, N. Origin and diversity of plant receptor-like kinases. Annu. Rev. Plant Biol. 2020, 71, 131–156. [Google Scholar] [CrossRef]
- Li, S.; Liu, Z.; Chen, G.; Qanmber, G.; Lu, L.; Zhang, J.; Ma, S.; Yang, Z.; Li, F. Identification and analysis of GhEXO gene family indicated that GhEXO7_At promotes plant growth and development through brassinosteroid signaling in cotton (Gossypium hirsutum L.). Front. Plant Sci. 2021, 12, 719889. [Google Scholar] [CrossRef]
- Šimura, J.; Antoniadi, I.; Široká, J.; Tarkowská, D.e.; Strnad, M.; Ljung, K.; Novák, O. Plant hormonomics: Multiple phytohormone profiling by targeted metabolomics. Plant Physiol. 2018, 177, 476–489. [Google Scholar] [CrossRef]
- Clouse, S.D. Brassinosteroid signal transduction: From receptor kinase activation to transcriptional networks regulating plant development. Plant Cell 2011, 23, 1219–1230. [Google Scholar] [CrossRef]
- Clouse, S.D.; Langford, M.; McMorris, T.C. A brassinosteroid-insensitive mutant in Arabidopsis thaliana exhibits multiple defects in growth and development. Plant Physiol. 1996, 111, 671–678. [Google Scholar] [CrossRef]
- Kim, E.-J.; Russinova, E. Brassinosteroid signalling. Curr. Biol. 2020, 30, R294–R298. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, L.P. IPC–isoelectric point calculator. Biol. Direct 2016, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of protein subcellular localization. Proteins Struct. Funct. Bioinform. 2006, 64, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Martelli, P.L.; Fariselli, P.; Profiti, G.; Casadio, R. BUSCA: An integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 2018, 46, W459–W466. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2. 0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Grigorova, B.; Vaseva, I.; Demirevska, K.; Feller, U. Combined Drought and Heat Stress in Wheat: Changes in Some Heat Shock Proteins. Biol. Plant. 2011, 55, 105–111. [Google Scholar] [CrossRef]
- Kim, D.K.; Kesawat, M.S.; Hong, C.B. One gene member of the ADP-ribosylation factor family is heat-inducible and enhances seed germination in Nicotiana tabacum. Genes Genom. 2017, 39, 1353–1365. [Google Scholar] [CrossRef]
- Kesawat, M.S.; Kim, D.K.; Zeba, N.; Suh, M.C.; Xia, X.; Hong, C.B. Ectopic RING zinc finger gene from hot pepper induces totally different genes in lettuce and tobacco. Mol. Breed. 2018, 38, 1–24. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Narancio, R.; John, U.; Mason, J.; Spangenberg, G. Selection of optimal reference genes for quantitative RT-PCR transcript abundance analysis in white clover (Trifolium repens L.). Funct. Plant Biol. 2018, 45, 737–744. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proposed Gene Name | Gene ID | Genomic Location | Orientation | CDS Length (bp) | Intron Number | Protein Length (aa) | Molecular Weight (KDa) | Isoelectric Point (pI) | GRAVY | Predicted Subcellular Localization |
|---|---|---|---|---|---|---|---|---|---|---|
| TaPERK1 | TraesCS1A02G127900 | 1A:155693812–155696618 | Forward | 1977 | 7 | 658 | 69.44 | 7.53 | −0.531 | Nucleus |
| TaPERK2 | TraesCS1B02G1470000 | 1B:209130189–209130266 | Reverse | 1431 | 8 | 476 | 52.14 | 6.17 | −0.5 | Nucleus |
| TaPERK3 | TraesCS1D02G00430 | 1D:2110107–2112027 | Forward | 1971 | 7 | 656 | 68.93 | 9.04 | −0.393 | Chloroplast outer membrane |
| TaPERK4 | TraesCS1D02G126300 | 1D:137437684–137440387 | Reverse | 1962 | 7 | 653 | 69.07 | 7.21 | −0.52 | Nucleus |
| TaPERK5 | TraesCS2A02G418200 | 2A:674030843–674031911 | Forward | 3048 | 23 | 1015 | 110.33 | 6.33 | −0.193 | Plasma membrane |
| TaPERK6 | TraesCS2A02G418300 | 2A:674050369–674051442 | Forward | 3042 | 23 | 1013 | 110.39 | 7.06 | −0.135 | Plasma membrane |
| TaPERK7 | TraesCS2A02G418400 | 2A:674061248–674062244 | Forward | 3159 | 23 | 1052 | 113.73 | 5.96 | −0.127 | Plasma membrane |
| TaPERK8 | TraesCS2B02G437200 | 2B:629023953–629025021 | Forward | 3045 | 23 | 1014 | 110.38 | 6.3 | −0.178 | Plasma membrane |
| TaPERK9 | TraesCS2B02G437300 | 2B:629106216–629107285 | Forward | 3048 | 23 | 1015 | 110.62 | 6.61 | −0.147 | Plasma membrane |
| TaPERK10 | TraesCS2D02G415600 | 2D:529537635–529538701 | Forward | 3048 | 22 | 1015 | 110.34 | 6.6 | −0.167 | Plasma membrane |
| TaPERK11 | TraesCS2D02G415700 | 2D:529548057–529548998 | Forward | 2775 | 23 | 924 | 101.07 | 7.29 | −0.182 | Plasma membrane |
| TaPERK12 | TraesCS2D02G415800 | 2D:529558487–529559547 | Forward | 3156 | 23 | 1051 | 113.45 | 6.11 | −0.102 | Plasma membrane |
| TaPERK13 | TraesCS3A02G003900 | 3A:1925607–1927275 | Reverse | 2064 | 7 | 687 | 72.42 | 5.96 | −0.429 | Plasma membrane |
| TaPERK14 | TraesCS3A02G152200 | 3A:142891955–142894634 | Forward | 1893 | 7 | 630 | 67.43 | 6.28 | −0.569 | Endomembrane system |
| TaPERK15 | TraesCS3A02G229800 | 3A:429615911–429617422 | Reverse | 2163 | 6 | 720 | 74.97 | 7.93 | −0.401 | Chloroplast thylakoid lumen |
| TaPERK16 | TraesCS3A02G278100 | 3A:507637093–507638935 | Reverse | 2028 | 7 | 675 | 72.42 | 7.31 | −0.481 | Plasma membrane |
| TaPERK17 | TraesCS3A02G290300 | 3A:519244808–519246110 | Reverse | 2184 | 7 | 727 | 75.8 | 6.11 | −0.535 | Endomembrane system |
| TaPERK18 | TraesCS3B02G008600 | 3B:4324660–4326408 | Forward | 2061 | 7 | 686 | 71.88 | 5.97 | −0.437 | Plasma membrane |
| TaPERK19 | TraesCS3B02G179300 | 3B:187347873–187350697 | Forward | 1896 | 7 | 631 | 67.46 | 6.35 | −0.569 | Endomembrane system |
| TaPERK20 | TraesCS3B02G259100 | 3B:416806224–416809608 | Reverse | 2097 | 6 | 698 | 72.98 | 7.63 | −0.448 | Plasma membrane |
| TaPERK21 | TraesCS3B02G312300 | 3B:501498044–501499926 | Reverse | 2034 | 7 | 677 | 72.6 | 7.31 | −0.493 | Endomembrane system |
| TaPERK22 | TraesCS3B02G325100 | 3B:525990462–525991846 | Reverse | 2436 | 7 | 811 | 84.77 | 6.09 | −0.51 | Plasma membrane |
| TaPERK23 | TraesCS3D02G005400 | 3D:2141185–2143272 | Forward | 1206 | 6 | 401 | 44.37 | 5.55 | −0.403 | Nucleus |
| TaPERK24 | TraesCS3D02G160000 | 3D:130928685–130931461 | Forward | 1899 | 7 | 632 | 67.49 | 6.36 | −0.555 | Endomembrane system |
| TaPERK25 | TraesCS3D02G278400 | 3D:385473929–385474240 | Reverse | 2031 | 8 | 676 | 72.56 | 7.1 | −0.471 | Endomembrane system |
| TaPERK26 | TraesCS3D02G290100 | 3D:400311470–400312883 | Reverse | 1317 | 6 | 438 | 47.14 | 5.97 | −0.447 | Nucleus |
| TaPERK27 | TraesCS4A02G077500 | 4A:76627667–76628358 | Forward | 1866 | 5 | 621 | 64.49 | 5.58 | −0.457 | Endomembrane system |
| TaPERK28 | TraesCS4A02G449700 | 4A:715718345–715719349 | Forward | 2604 | 19 | 867 | 94.75 | 7.03 | −0.145 | Plasma membrane |
| TaPERK29 | TraesCS4B02G233600 | 4B:486206279–486206961 | Reverse | 1857 | 5 | 618 | 64.45 | 5.63 | −0.487 | Plasma membrane |
| TaPERK30 | TraesCS5A02G411300 | 5A:599978835–599979642 | Reverse | 1722 | 4 | 573 | 60.33 | 7.96 | −0.387 | Chloroplast outer membrane |
| TaPERK31 | TraesCS5B02G415000 | 5B:589228532–589228944 | Reverse | 1842 | 3 | 613 | 64.68 | 7.86 | −0.368 | Chloroplast outer membrane |
| TaPERK32 | TraesCS7A02G038600 | 7A:17358644–17359648 | Reverse | 3030 | 23 | 1009 | 109.7 | 6.22 | −0.108 | Plasma membrane |
| TaPERK33 | TraesCS7A02G231900 | 7A:202852283–202853761 | Reverse | 2187 | 6 | 728 | 76.24 | 5.32 | −0.484 | Nucleus |
| TaPERK34 | TraesCS7B02G130400 | 7B:156752944–156754400 | Reverse | 2208 | 6 | 735 | 76.89 | 5.22 | −0.49 | Nucleus |
| TaPERK35 | TraesCS7D02G034800 | 7D:17864178–17865182 | Reverse | 3021 | 23 | 1006 | 109.26 | 6.1 | −0.09 | Plasma membrane |
| TaPERK36 | TraesCS7D02G232700 | 7D:194224547–194225929 | Forward | 2256 | 6 | 751 | 78.2 | 6.16 | −0.645 | Endomembrane system |
| TaPERK37 | TraesCSU02G104700 | Un:92294980–92296477 | Reverse | 2205 | 6 | 734 | 76.55 | 5.32 | −0.483 | Nucleus |
| Plant Species | Genome Size (Approx.) | Coding Genes | PERK Genes |
|---|---|---|---|
| Triticum aestivum (6n) | 17 Gb | 107,891 | 37 |
| Arabidopsis thaliana (2n) | 135 Mb | 27,655 | 15 |
| Oryza sativa | 500 Mb | 37,960 | 8 |
| Zea mays (2n) | 2.4 Gb | 39,591 | 23 |
| Glycine max (2n) | 1.15 Gb | 55,897 | 16 |
| Sorghum bicolor (2n) | 730 Mb | 28,120 | 15 |
| Gossypium arboretum (2n) | 1746 Mb | 41,330 | 15 |
| Gossypium raimondii (2n) | 885 Mb | 40,976 | 16 |
| Gossypium hirsutum (4n) | 2.43 Gb | 75,376 | 33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kesawat, M.S.; Kherawat, B.S.; Singh, A.; Dey, P.; Routray, S.; Mohapatra, C.; Saha, D.; Ram, C.; Siddique, K.H.M.; Kumar, A.; et al. Genome-Wide Analysis and Characterization of the Proline-Rich Extensin-like Receptor Kinases (PERKs) Gene Family Reveals Their Role in Different Developmental Stages and Stress Conditions in Wheat (Triticum aestivum L.). Plants 2022, 11, 496. https://doi.org/10.3390/plants11040496
Kesawat MS, Kherawat BS, Singh A, Dey P, Routray S, Mohapatra C, Saha D, Ram C, Siddique KHM, Kumar A, et al. Genome-Wide Analysis and Characterization of the Proline-Rich Extensin-like Receptor Kinases (PERKs) Gene Family Reveals Their Role in Different Developmental Stages and Stress Conditions in Wheat (Triticum aestivum L.). Plants. 2022; 11(4):496. https://doi.org/10.3390/plants11040496
Chicago/Turabian StyleKesawat, Mahipal Singh, Bhagwat Singh Kherawat, Anupama Singh, Prajjal Dey, Snehasish Routray, Chinmayee Mohapatra, Debanjana Saha, Chet Ram, Kadambot H. M. Siddique, Ajay Kumar, and et al. 2022. "Genome-Wide Analysis and Characterization of the Proline-Rich Extensin-like Receptor Kinases (PERKs) Gene Family Reveals Their Role in Different Developmental Stages and Stress Conditions in Wheat (Triticum aestivum L.)" Plants 11, no. 4: 496. https://doi.org/10.3390/plants11040496
APA StyleKesawat, M. S., Kherawat, B. S., Singh, A., Dey, P., Routray, S., Mohapatra, C., Saha, D., Ram, C., Siddique, K. H. M., Kumar, A., Gupta, R., Chung, S.-M., & Kumar, M. (2022). Genome-Wide Analysis and Characterization of the Proline-Rich Extensin-like Receptor Kinases (PERKs) Gene Family Reveals Their Role in Different Developmental Stages and Stress Conditions in Wheat (Triticum aestivum L.). Plants, 11(4), 496. https://doi.org/10.3390/plants11040496