
Genetic Diversity and Population Structure of Chinese Chestnut (Castanea mollissima Blume) Cultivars Revealed by GBS Resequencing
 ,
,
Abstract
1. Introduction
2. Material and Methods
2.1. Plant Material
2.2. Sequencing and Mapping
2.3. SNP Detection and Annotation
2.4. Population Stratification Analysis
3. Results
3.1. Sequencing Data Analysis
3.2. Genetic Diversity Analysis of Population
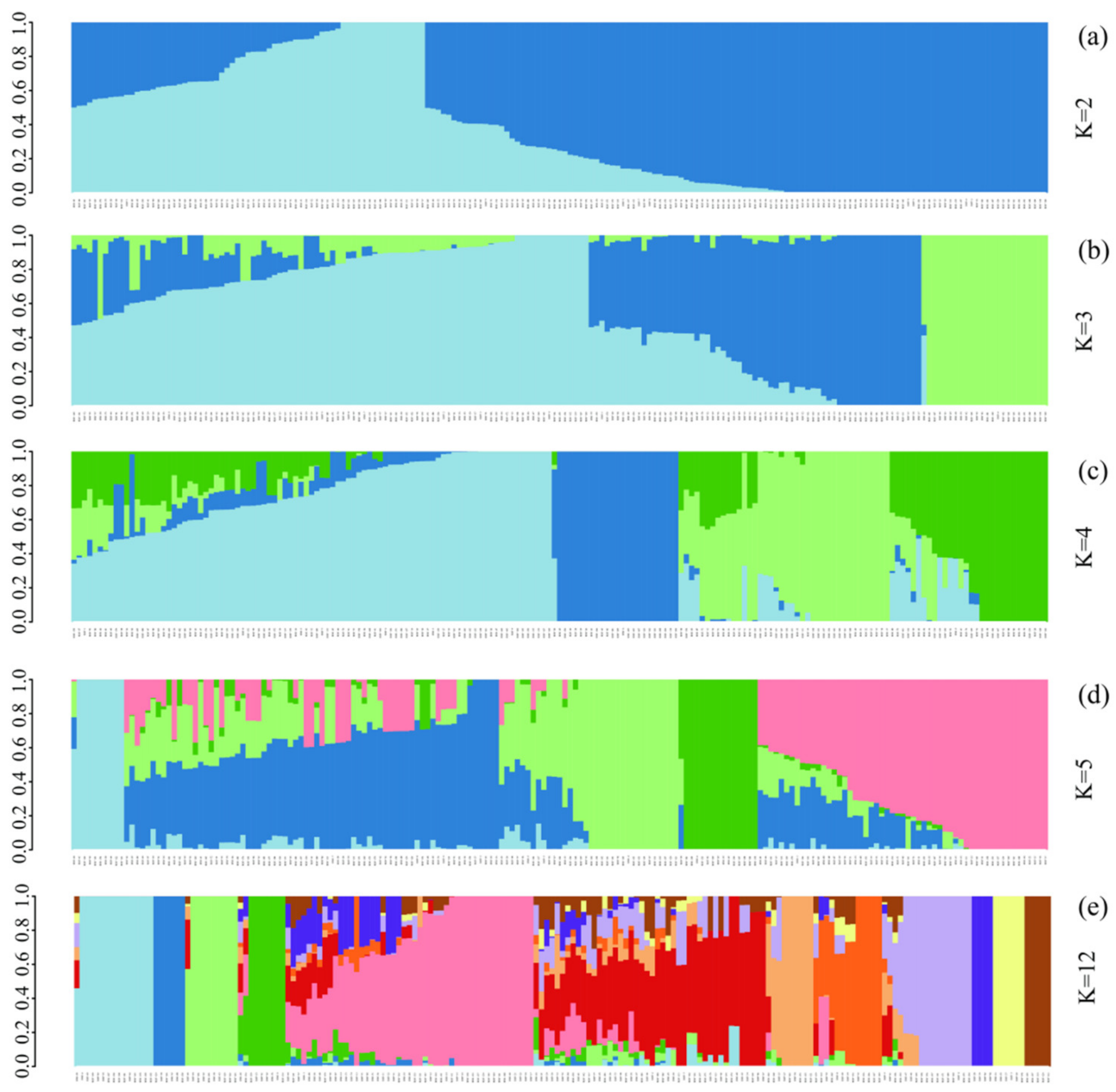
3.3. ADMIXTURE Analysis of 185 Chinese Chestnut Landraces
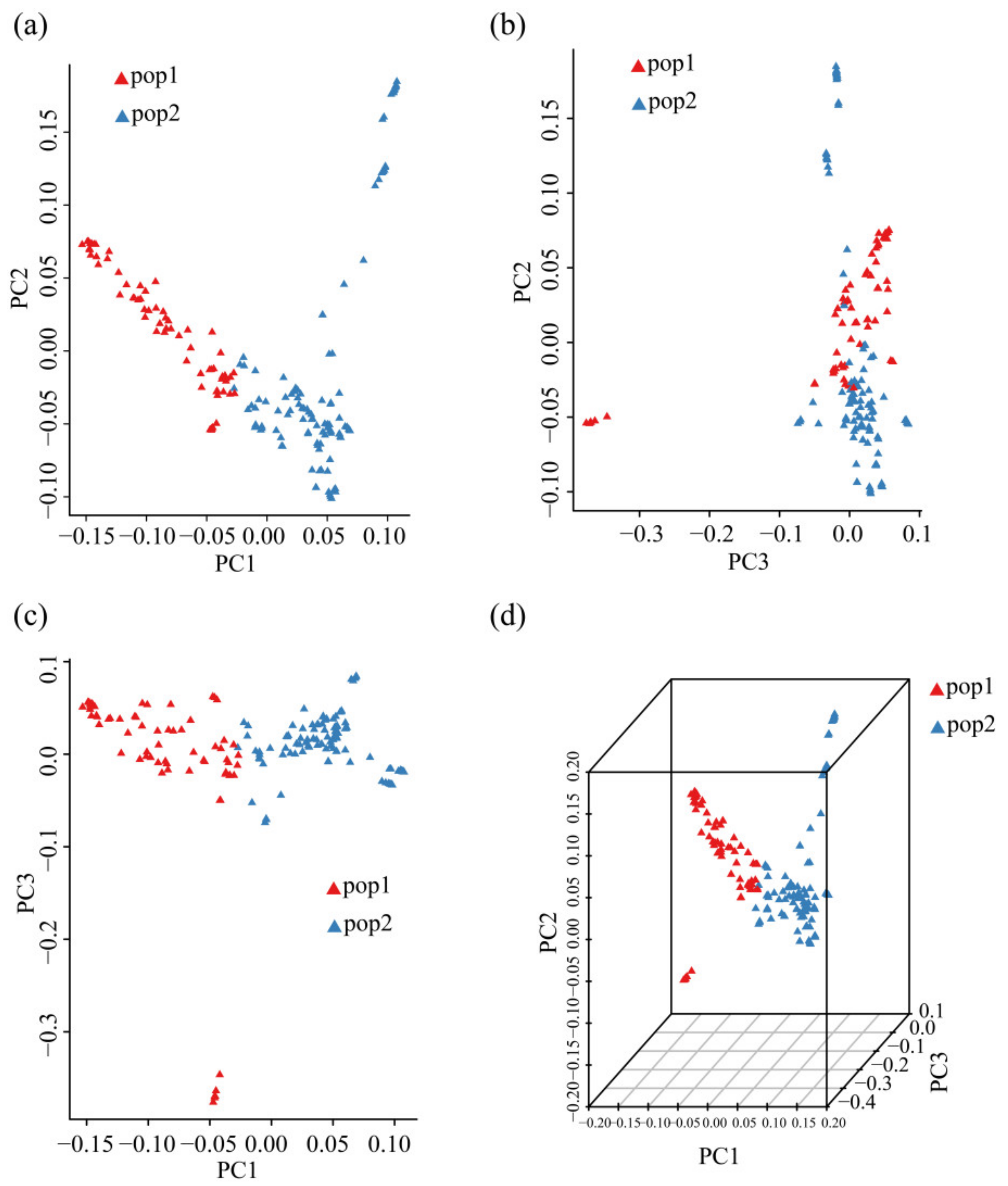
3.4. PCA of 185 Chinese Chestnut Landraces
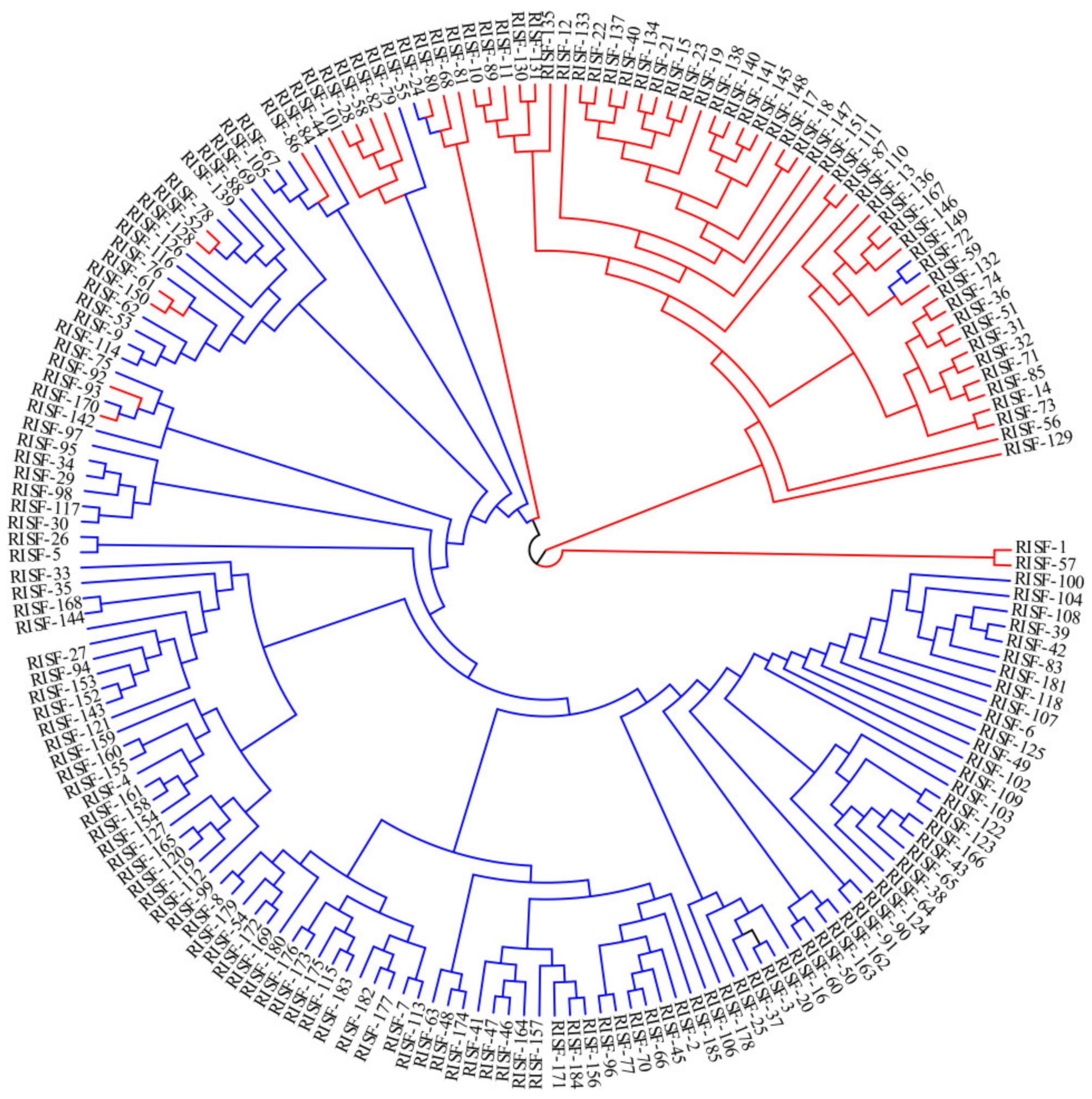
3.5. Phylogenetic Analysis of 185 Chinese Chestnut Landraces
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, S.; Wang, L.T.; Fu, Y.J.; Jiang, J.C. Bioactive constituents, nutritional benefits and woody food applications of Castanea mollissima: A comprehensive review. Food Chem. 2022, 393, 133380. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Jiang, G.; Gu, C.; Yang, H.; Jiang, Y. Structural changes in polysaccharides isolated from chestnut (Castanea mollissima Bl.) fruit at different degrees of hardening. Food Chem. 2010, 119, 1211–1215. [Google Scholar]
- Brewer, L.G. Ecology of Survival and Recovery from Blight in American Chestnut Trees (Castanea dentata (Marsh.) Borkh.) in Michigan. Bull. Torrey Bot. Club 1995, 122, 40–57. [Google Scholar] [CrossRef]
- Anagnostakis, S.L. Chestnut blight: The classical problem of an introduced pathogen. Mycologia 1987, 79, 23–37. [Google Scholar] [CrossRef]
- De Vasconcelos, M.D.C.B.M.; Bennett, R.N.; Rosa, E.A.S.; Cardoso, J.V.F. Primary and Secondary Metabolite Composition of Kernels from Three Cultivars of Portuguese Chestnut (Castanea sativa Mill.) at Different Stages of Industrial Transformation. J. Agric. Food Chem. 2007, 55, 3508–3516. [Google Scholar] [CrossRef]
- Rutter, P.; Miller, G.; Payne, J. Chestnuts. In Genetic Resources of Temperate Fruit and Nut Crops; Moore, J.N., Ballington, J.R., Jr., Eds.; The International Society for Horticultural Science: Wageningen, The Netherlands, 1990; pp. 761–788. [Google Scholar]
- Zohary, D.; Hopf, M.; Weiss, E. Domestication of Plants in the Old World: The Origin and Spread of Domesticated Plants in Southwest Asia, Europe, and the Mediterranean Basin; Oxford University Press: Oxford, UK, 2012; pp. 220–227. [Google Scholar]
- LaBonte, N.R.; Zhao, P.; Woeste, K. Signatures of Selection in the Genomes of Chinese Chestnut (Castanea mollissima Blume): The Roots of Nut Tree Domestication. Front. Plant Sci. 2018, 9, 810. [Google Scholar] [CrossRef]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef]
- Ong, Q.; Nguyen, P.; Thao, N.P.; Le, L. Bioinformatics Approach in Plant Genomic Research. Curr. Genom. 2016, 17, 368–378. [Google Scholar] [CrossRef]
- Jiang, X.B.; Tang, D.; Gong, B.C. Genetic diversity and association analysis of Chinese chestnut (Castanea mollissima Blume) cultivars based on SSR markers. Braz. J. Bot. 2017, 40, 235–246. [Google Scholar] [CrossRef]
- Tang, R.; Liu, E.X.; Zhang, Y.Z.; Schinnerl, J.; Sun, W.B.; Chen, G. Genetic diversity and population structure of Amorphophallus albus, a plant species with extremely small populations (PSESP) endemic to dry-hot valley of Jinsha River. BMC Genet. 2020, 21, 102. [Google Scholar] [CrossRef]
- Unamba, C.I.N.; Nag, A.; Sharma, R.K. Next Generation Sequencing Technologies: The Doorway to the Unexplored Genomics of Non-Model Plants. Front. Plant Sci. 2015, 6, 1074. [Google Scholar] [CrossRef] [PubMed]
- Rayda, B.A.; Fabienne, M.; Hajer, B.H.; Ahmed, R.; Sezi, E.; Narendra, K.; Mohsen, H.; Amine, A.; Riaz, U.; Essam, A.A. SNP discovery and structural insights into OeFAD2 unravelling high oleic/linoleic ratio in olive oil. Comput. Struct. Biotechnol. J. 2022, 20, 1229–1243. [Google Scholar]
- Liu, G.; Xie, Y.J.; Zhang, D.Q.; Chen, H.P. Analysis of SSR loci and development of SSR primers in Eucalyptus. J. For. Res. 2018, 29, 273–282. [Google Scholar] [CrossRef]
- Li, Y.F.; Zhang, J.J.; Chang, S.X.; Jiang, P.K.; Zhou, G.M.; Shen, Z.M.; Wu, J.S.; Lin, L.; Wang, Z.S.; Shen, M.C. Converting native shrub forests to Chinese chestnut plantations and subsequent intensive management affected soil C and N pools. For. Ecol. Manag. 2014, 312, 161–169. [Google Scholar] [CrossRef]
- Liu, Y.L.; Li, S.Q.; Wang, Y.Y.; Liu, P.Y.; Han, W.J. De novo assembly of the seed transcriptome and search for potential EST-SSR markers for an endangered, economically important tree species: Elaeagnus mollis Diels. J. For. Res. 2019, 31, 759–767. [Google Scholar] [CrossRef]
- Taranto, F.; D’Agostino, N.; Greco, B.; Cardi, T.; Tripodi, P. Genome-wide SNP discovery and population structure analysis in pepper (Capsicum annuum) using genotyping by sequencing. BMC Genom. 2016, 17, 943. [Google Scholar] [CrossRef]
- Nunziata, A.; Ruggieri, V.; Petriccione, M.; De Masi, L. Single Nucleotide Polymorphisms as Practical Molecular Tools to Support European Chestnut Agrobiodiversity Management. Int. J. Mol. Sci. 2020, 21, 4805. [Google Scholar] [CrossRef]
- Nunziata, A.; Ferlito, F.; Magri, A.; Ferrara, E.; Petriccione, M. The Hundred Horses Chestnut: A model system for studying mutation rate during clonal propagation in superior plants. Forestry 2022, 95, 678–685. [Google Scholar] [CrossRef]
- Chang, L.; Wang, S.; Chang, X.; Wang, S.J.F.H. Structural and functional properties of starches from Chinese chestnuts. Food Hydrocoll. 2015, 43, 568–576. [Google Scholar]
- He, J.F.; Zhao, Q.X.; Laroche, A.; Lu, Z.X.; Liu, H.K.; Li, Z.Q. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5, 484. [Google Scholar] [CrossRef]
- Nunzio, D.A.; Francesca, T.; Salvatore, C.; Giacomo, M.; Valentina, F.; Susanna, G.; Monica, M.M.; Stefano, P.; di Valentina, R.; Wilma, S.; et al. GBS-derived SNP catalogue unveiled wide genetic variability and geographical relationships of Italian olive cultivars. Sci. Rep. 2018, 8, 15877. [Google Scholar]
- Jiří, K.; Jaroslav, Č.; Jan, S.; Zuzana, F.; Jakub, D.; Milan, L.; Yousry, A. Genetic diversity of Norway spruce ecotypes assessed by GBS-derived SNPs. Sci. Rep. 2021, 11, 23119. [Google Scholar]
- Ji, F.; Wei, W.; Liu, Y.; Wang, G.; Zhang, Q.; Xing, Y.; Zhang, S.; Liu, Z.; Cao, Q.; Qin, L. Construction of a SNP-Based High-Density Genetic Map Using Genotyping by Sequencing (GBS) and QTL Analysis of Nut Traits in Chinese Chestnut (Castanea mollissima Blume). Front. Plant Sci. 2018, 9, 816. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- García-Arias, F.L.; Osorio-Guarín, J.A.; Zarantes, V.M.N. Association Study Reveals Novel Genes Related to Yield and Quality of Fruit in Cape Gooseberry (Physalis peruviana L.). Front. Plant Sci. 2018, 9, 362. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef]
- Ai, C.X.; Li, G.T.; Zhang, L.S.; Liu, Q.Z. Study on the genetic diversity of natural chestnut populations in Shandong China by SSR markers. Acta Hortic. 2009, 844, 257–266. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Hohenlohe, P.A.; Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 2010, 11, 697–709. [Google Scholar] [CrossRef]
- Lusini, I.; Velichkov, I.; Pollegioni, P.; Chiocchini, F.; Hinkov, G.; Zlatanov, T.; Cherubini, M.; Mattioni, C. Estimating the genetic diversity and spatial structure of Bulgarian Castanea sativa populations by SSRs: Implications for conservation. Conserv. Genet. 2014, 15, 283–293. [Google Scholar] [CrossRef]
- Hao, H.N.; Li, Q.; Bao, W.J.; Wu, Y.W.; Ouyang, J. Relationship between physicochemical characteristics and in vitro digestibility of chestnut (Castanea mollissima) starch. Food Hydrocoll. 2018, 84, 193–199. [Google Scholar] [CrossRef]
- Aravanopoulos, F.A.; Jarman, R.; Mattioni, C.; Russell, K.; Chambers, F.M.; Bartlett, D.; Martin, M.A.; Cherubini, M.; Villani, F.; Webb, J. DNA analysis of Castanea sativa (sweet chestnut) in Britain and Ireland: Elucidating European origins and genepool diversity. PLoS ONE 2019, 14, e0222936. [Google Scholar] [CrossRef]
- Chen, X.N.; Zhang, B.; Chen, Y.J.; Hou, X.; Wang, J.; Chang, G. Effect of forest rodents on predation and dispersal of Castanea mollissima and Quercus aliena seeds on south-and north-facing slopes of Qinling Mountains. Acta Ecol. Sin. 2016, 36, 1303–1311. [Google Scholar] [CrossRef]
- Cao, L.; Xiao, Z.S.; Guo, C.; Chen, J.; Zhang, Z.B. Scatter-hoarding rodents as secondary seed dispersers of a frugivore-dispersed tree Scleropyrum wallichianum in a defaunated Xishuangbanna tropical forest. China. Integr. Zool. 2011, 6, 227–234. [Google Scholar] [CrossRef]
- Zhang, C.M.; Huang, J.; Yin, X.; Lian, C.L.; Li, X.G. Genetic diversity and population structure of sour jujube, Ziziphus acidojujuba. Tree Genet. Genomes 2014, 11, 809. [Google Scholar] [CrossRef]
- Feng, X.J.; Zhou, H.J.; Zulfiqar, S.; Luo, X.; Hu, Y.; Feng, L.; Malvolti, M.E.; Woeste, K.; Zhao, P. The Phytogeographic History of Common Walnut in China. Front. Plant Sci. 2018, 9, 1399. [Google Scholar] [CrossRef]
- Bouffartigue, C.; Debille, S.; Fabreguettes, O.; Cabrer, A.R.; Lorenzo, S.P.; Flutre, T.; Harvengt, L. Two main genetic clusters with high admixture between forest and cultivated chestnut (Castanea sativa Mill.) in France. Ann. For. Sci. 2020, 77, 74. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Parameter | Raw Base (bp) | Clean Base (bp) | Effective Rate (%) | Error Rate (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|
| Minimum | 89,598,816 | 89,598,816 | 100 | 0.02 | 88.27 | 81.43 | 33.47 |
| Maximum | 631,849,824 | 631,849,824 | 100 | 0.04 | 95.97 | 93.05 | 39.27 |
| Mean | 411,594,134 | 411,591,572 | 100 | 0.03 | 92.88 | 89.15 | 36.58 |
| Total | 76,246,654,464 | 76,246,175,232 |
| Parameter | Clean Reads | Mapped Reads | Mapping Rate (%) | Average Depth (X) | Coverage at Least 1X (%) | Coverage at Least 4X (%) |
|---|---|---|---|---|---|---|
| Minimum | 622,214 | 601,909 | 91.00 | 6.51 | 5.08 | 0.80 |
| Maximum | 4,387,846 | 4,227,193 | 98.13 | 14.75 | 19.20 | 6.13 |
| Mean | 2,858,275 | 2,769,234 | 96.91 | 9.74 | 11.80 | 4.32 |
| Category | Number of SNPs | |
|---|---|---|
| Total | 299,015 | |
| Upstream | 4065 | |
| Exonic | Stop gain | 209 |
| Stop loss | 9 | |
| Non-synonymous | 3800 | |
| Synonymous | 2241 | |
| Intronic | 4724 | |
| Splicing | 47 | |
| Downstream | 4449 | |
| Upstream/Downstream | 119 | |
| Intergenic | 277,994 | |
| Transitions (ts) | 204,498 | |
| Transversions (tv) | 94,517 | |
| ts/tv | 2.163 | |
| Population | Observed Heterozygosity (Ho) | Expected Heterozygosity (He) | Gene Flow (Nm) |
|---|---|---|---|
| North of the Yellow River | 0.27963 | 0.30531 | 1.7655 |
| Eastern Coastal region | 0.30334 | 0.30612 | 1.9817 |
| Yangtze River Basin region | 0.29995 | 0.30800 | 3.2154 |
| South Central region | 0.31340 | 0.30824 | 2.2518 |
| Midwest region | 0.34265 | 0.33958 | 1.4372 |
| Source of Variation | df | Sum Squares | Variance of Components | Percentage of Variation (%) |
|---|---|---|---|---|
| Among populations | 4 | 3879.027 | 8.75943 | 2.07 |
| Within populations | 365 | 151,380.206 | 414.74029 | 97.93 |
| Total | 369 | 155,259.232 | 423.49972 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, X.; Fang, Z.; Lai, J.; Wu, Q.; Wu, J.; Gong, B.; Wang, Y. Genetic Diversity and Population Structure of Chinese Chestnut (Castanea mollissima Blume) Cultivars Revealed by GBS Resequencing. Plants 2022, 11, 3524. https://doi.org/10.3390/plants11243524
Jiang X, Fang Z, Lai J, Wu Q, Wu J, Gong B, Wang Y. Genetic Diversity and Population Structure of Chinese Chestnut (Castanea mollissima Blume) Cultivars Revealed by GBS Resequencing. Plants. 2022; 11(24):3524. https://doi.org/10.3390/plants11243524
Chicago/Turabian StyleJiang, Xibing, Zhou Fang, Junsheng Lai, Qiang Wu, Jian Wu, Bangchu Gong, and Yanpeng Wang. 2022. "Genetic Diversity and Population Structure of Chinese Chestnut (Castanea mollissima Blume) Cultivars Revealed by GBS Resequencing" Plants 11, no. 24: 3524. https://doi.org/10.3390/plants11243524
APA StyleJiang, X., Fang, Z., Lai, J., Wu, Q., Wu, J., Gong, B., & Wang, Y. (2022). Genetic Diversity and Population Structure of Chinese Chestnut (Castanea mollissima Blume) Cultivars Revealed by GBS Resequencing. Plants, 11(24), 3524. https://doi.org/10.3390/plants11243524