
Chemical Modification of Curcumin into Its Semi-Synthetic Analogs Bearing Pyrimidinone Moiety as Anticancer Agents

 , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Isolation of Curcumin
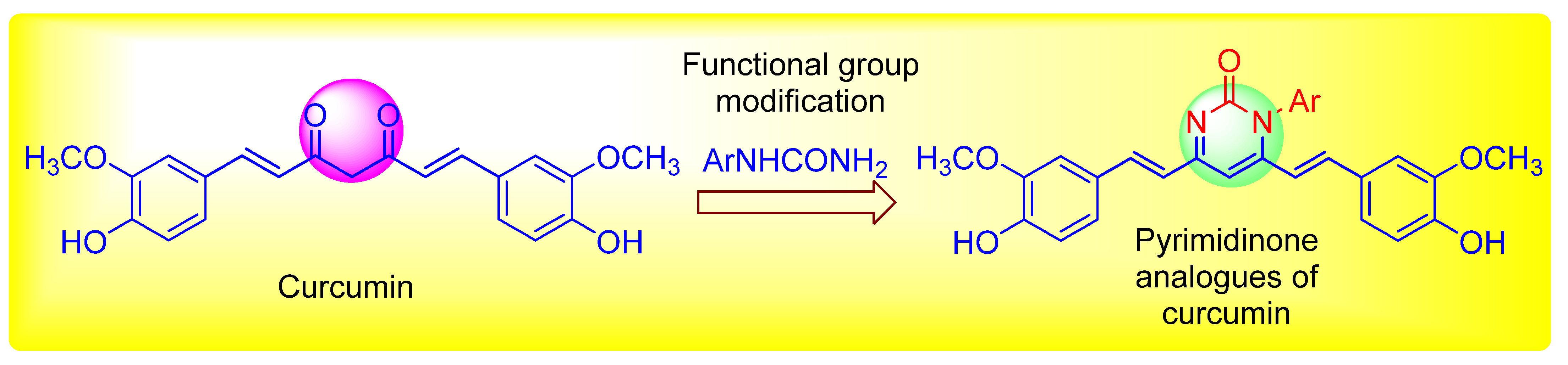
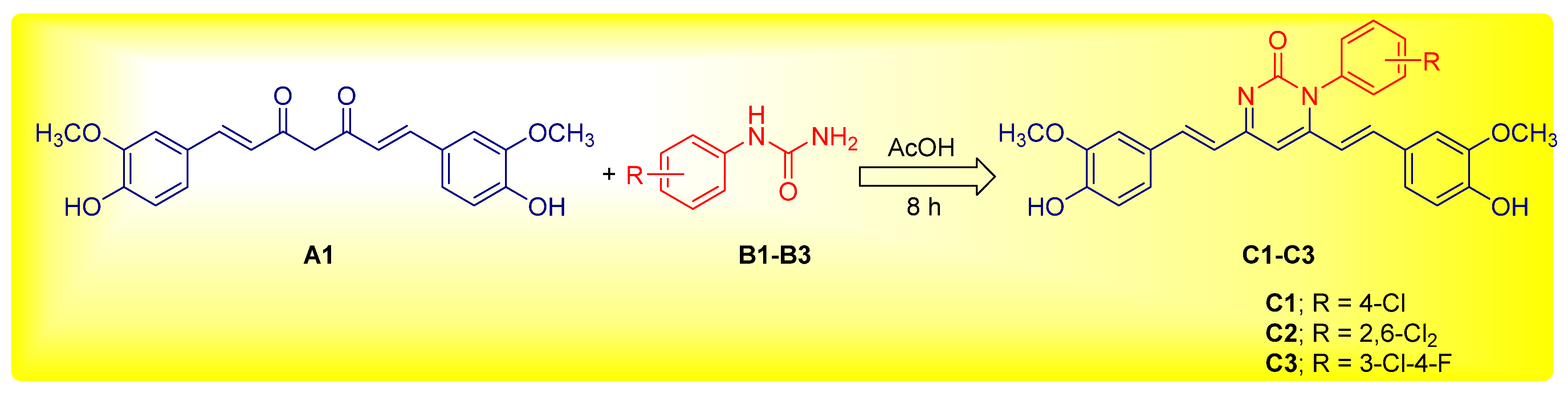
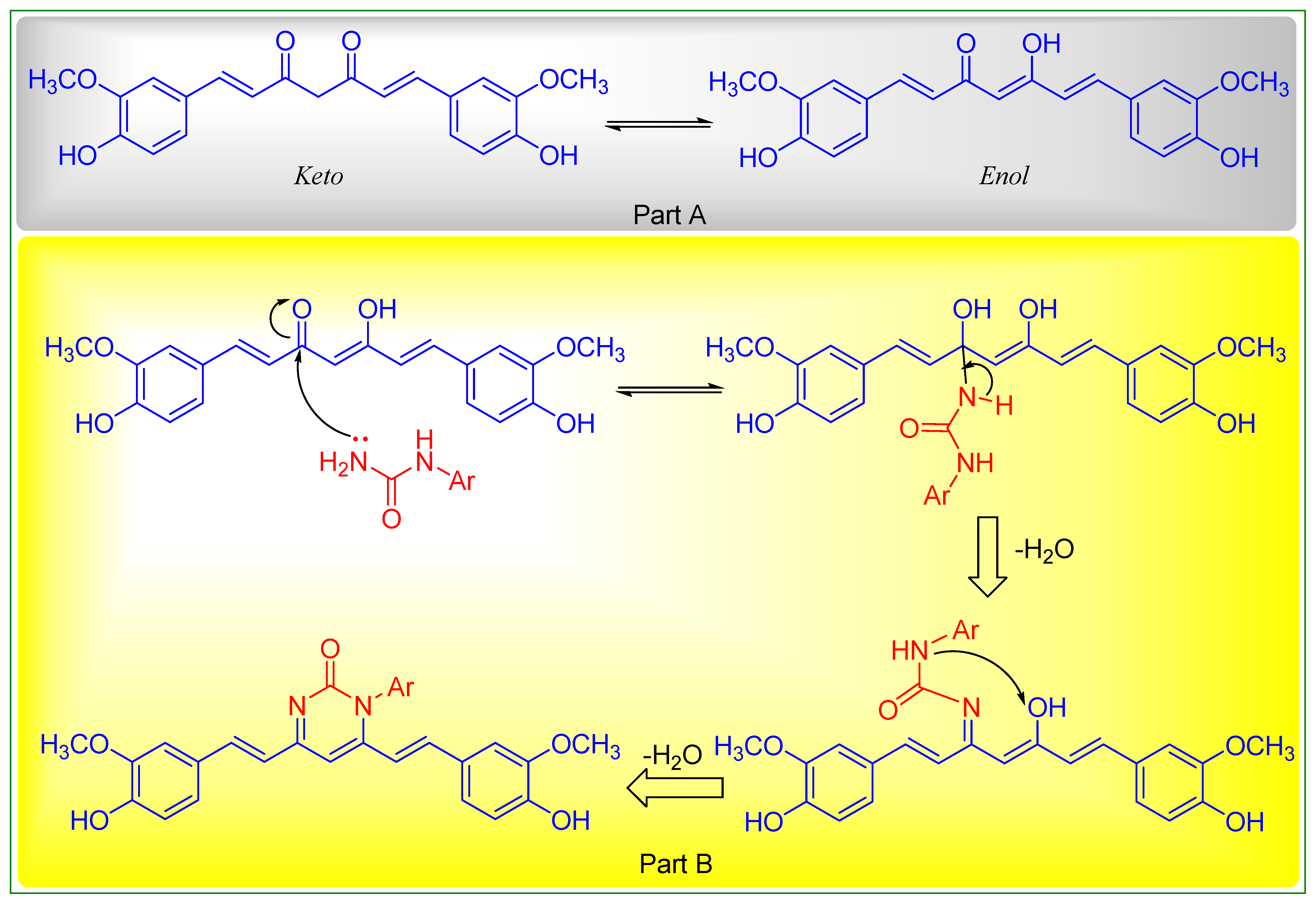
2.2. Chemistry
Preparation of 1-aryl-4,6-bis((E)-4-hydroxy-3-methoxystyryl)pyrimidin-2(1H)-one (C1-C3)
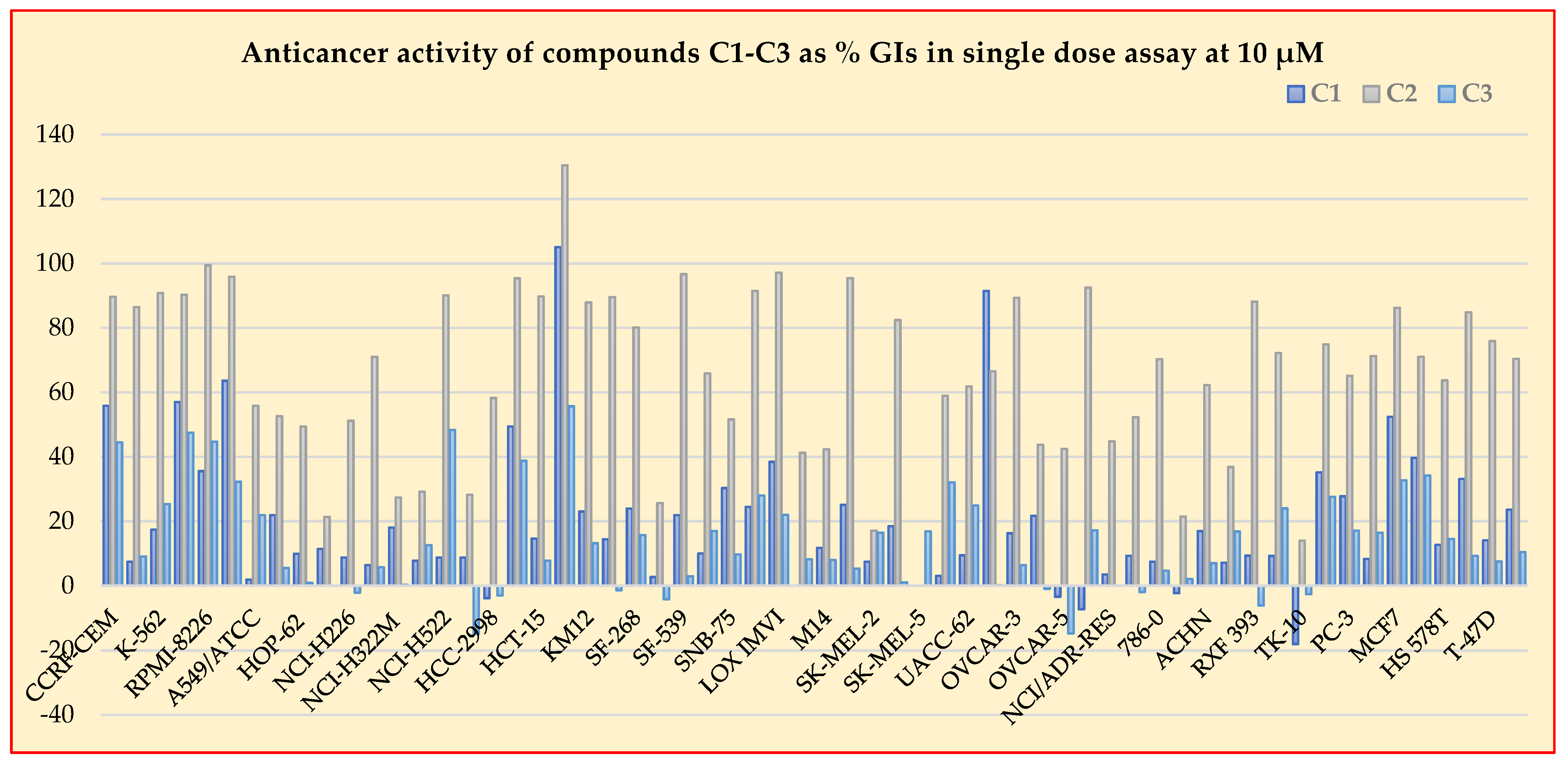
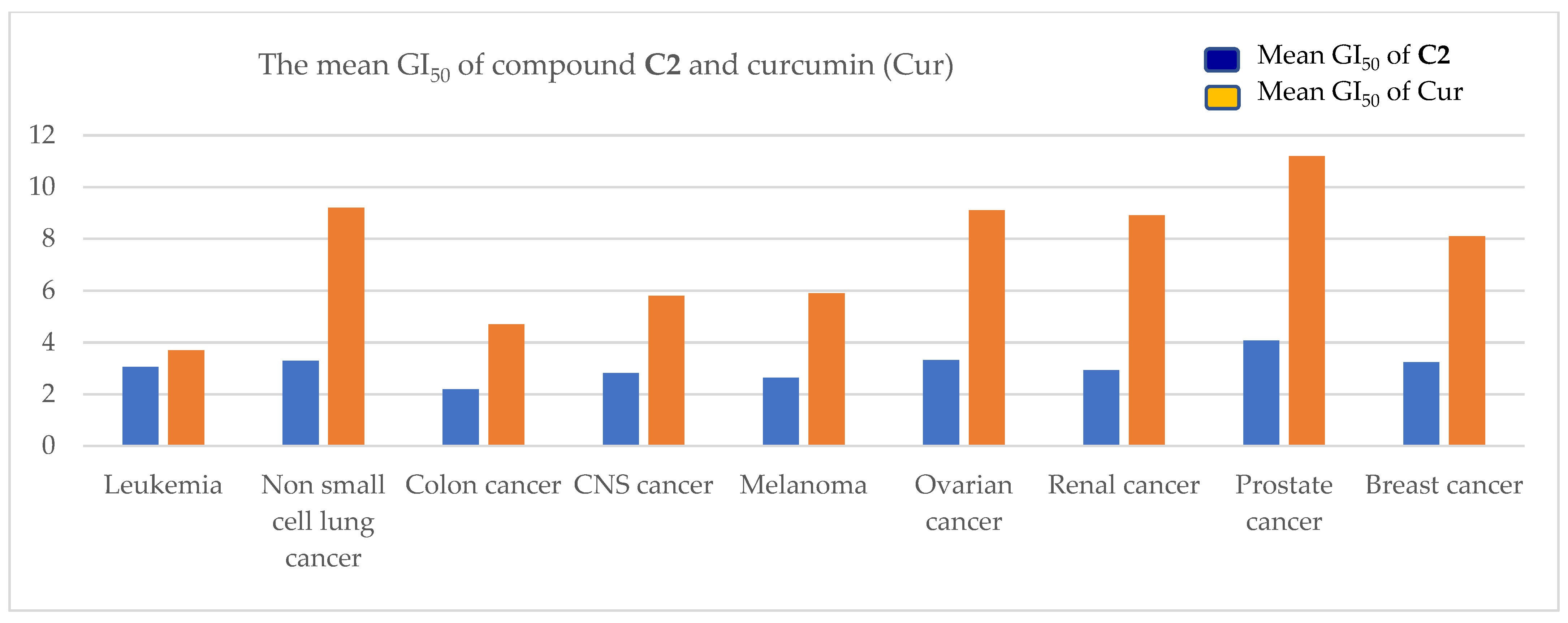
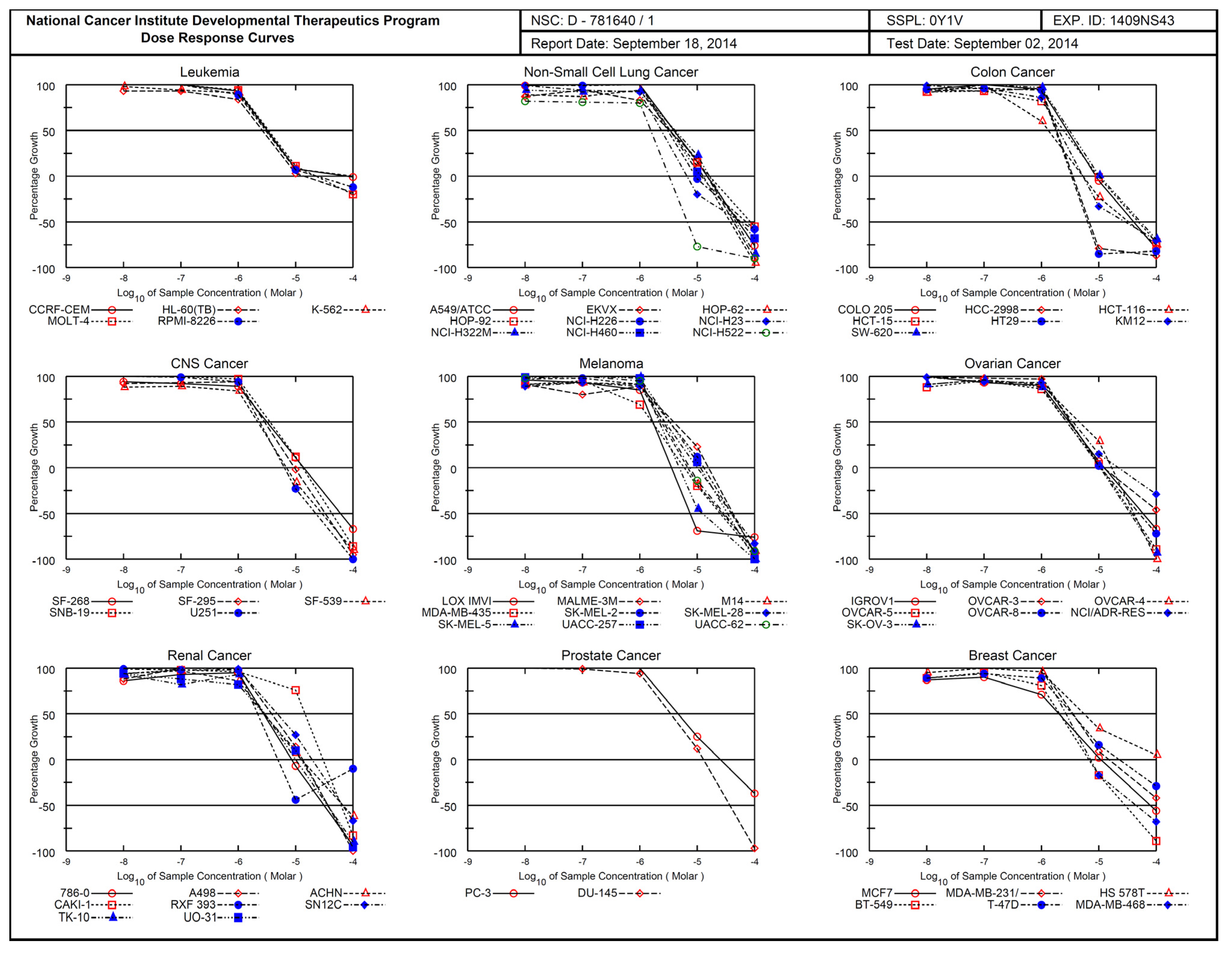
2.3. Antiproliferative Activity
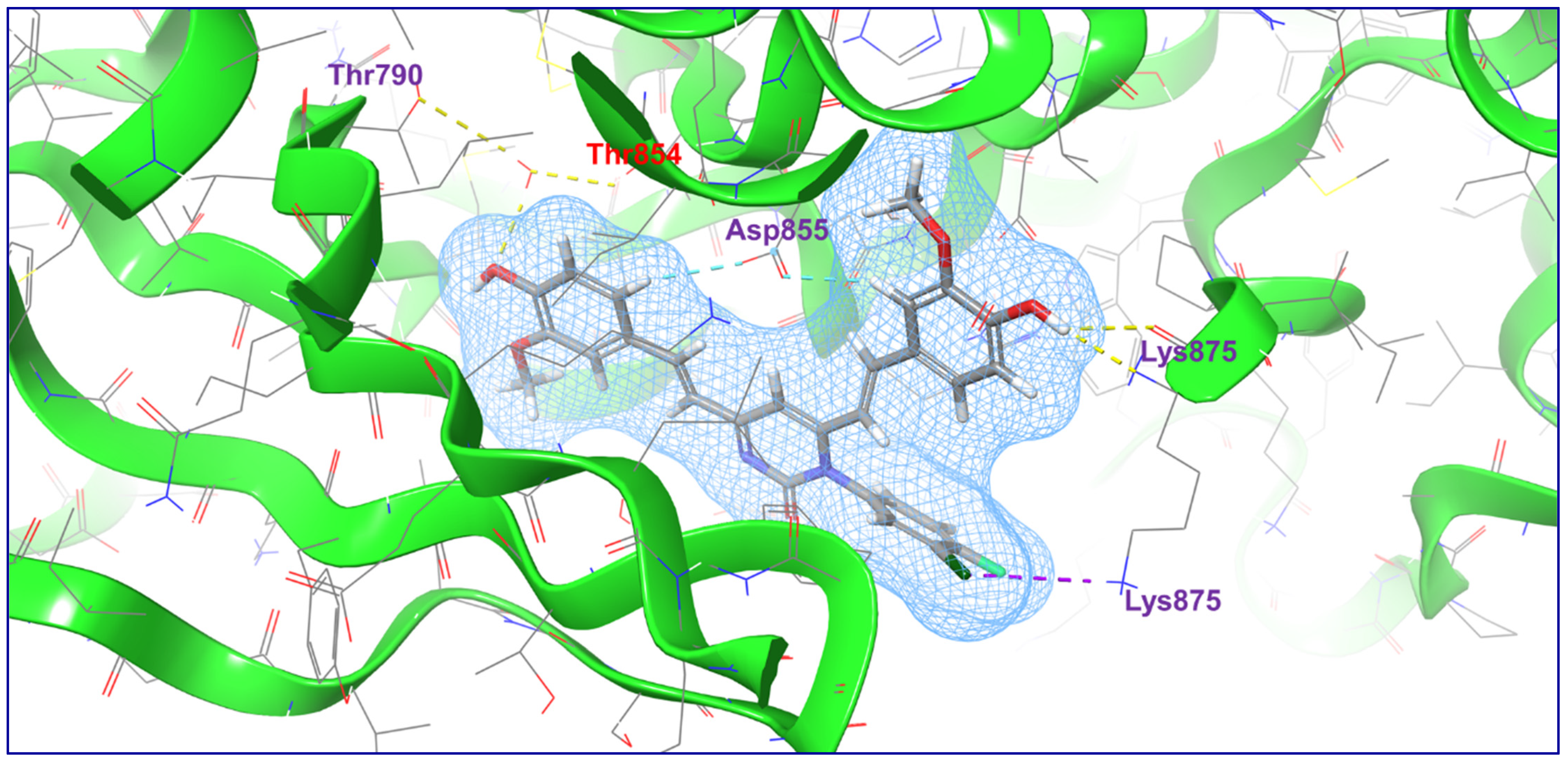
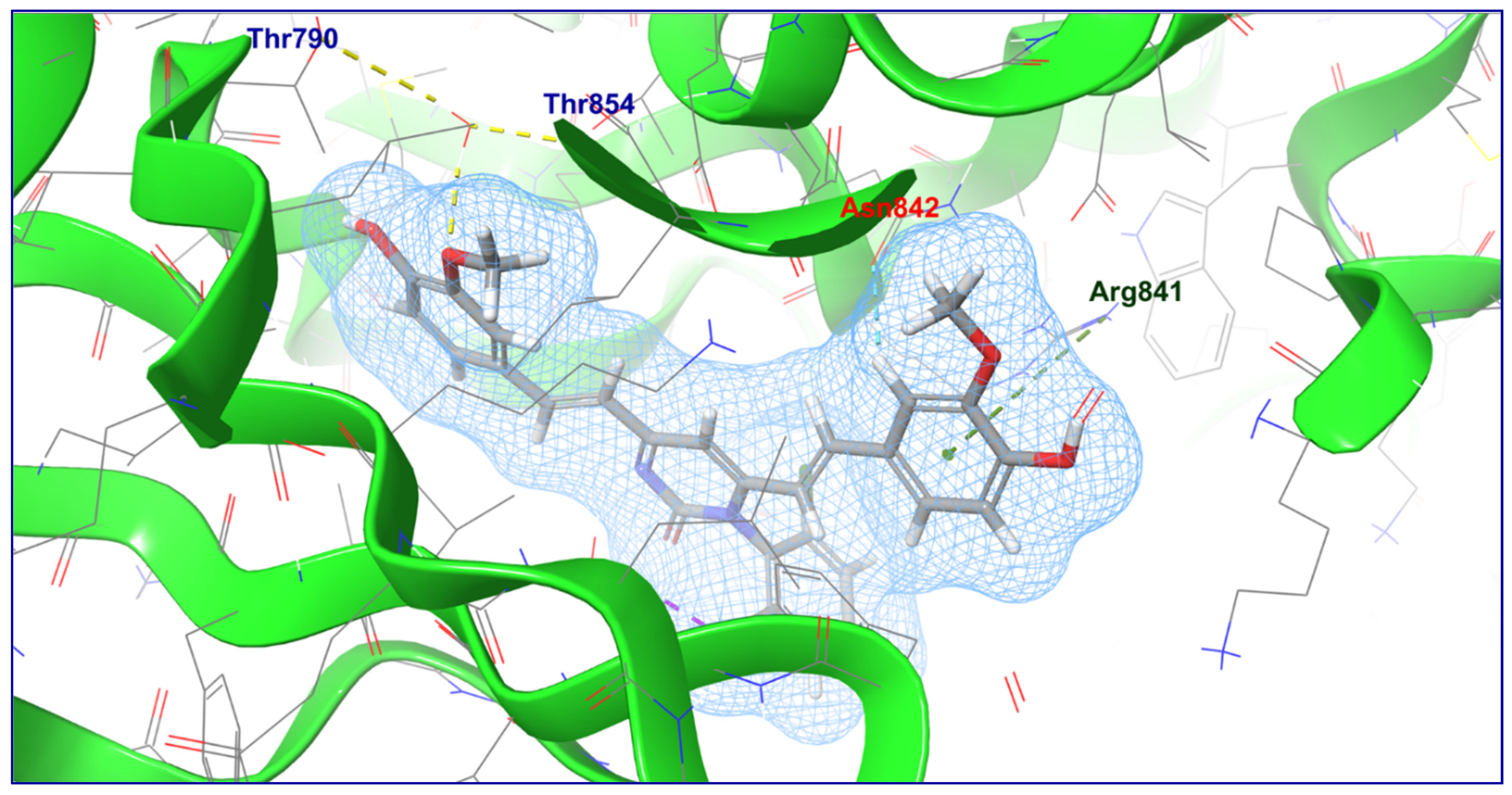
2.4. Molecular Docking Studies
3. Discussion
4. Materials and Methods
4.1. Preparation of Curcumin Analogue C1-C3
4.1.1. 1-(4-Chlorophenyl)-4,6-bis((E)-4-hydroxy-3-methoxystyryl)pyrimidin-2(1H)-one
4.1.2. 1-(2,6-Dichlorophenyl)-4,6-bis((E)-4-hydroxy-3-methoxystyryl)pyrimidin-2(1H)-one
4.1.3. 1-(3-Chloro-4-fluorophenyl)-4,6-bis((E)-4-hydroxy-3-methoxystyryl)pyrimidin-2(1H)-one
4.2. Anticancer Activity
4.3. Molecular Docking Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Ali, A.; Tahir, A.; Bakht, M.A.; Salahuddin; Ahsan, M.J. Molecular Engineering of Curcumin, an Active Constituent of Curcuma longa L. (Turmeric) of the Family Zingiberaceae with Improved Antiproliferative Activity. Plants 2021, 10, 1559. [Google Scholar] [CrossRef]
- Mishra, B.B.; Tiwari, V.K. Natural products: An evolving role in future drug discovery. Eur. J. Med. Chem. 2011, 46, 4769–4807. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Conlin, A.; de Azambuja, E.; Lago, L.D. Current perspectives of epothilones in breast cancer. Eur. J. Cancer 2008, 44, 341–352. [Google Scholar]
- Grossman, S.A.; Carson, K.A.; Phuphanich, S.; Batchelor, T.; Peereboom, D.; Nabors, L.B.; Lesser, G.; Hausheer, F. Phase I and pharmacokinetic study of karenitecin in patients with recurrent malignant gliomas. Neuro-oncology 2008, 10, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S. Natural products to drugs: Natural product-derived compounds in clinical trials. Nat. Prod. Rep. 2008, 25, 475. [Google Scholar] [CrossRef]
- Sessa, C.; Cresta, S.; Cerny, T.; Baselga, J.; Caremoli, E.R.; Malossi, A.; Hess, D.; Trigo, J.; Zucchetti, M.; D’Incalci, M.; et al. Concerted escalation of dose and dosing duration in a phase I study of the oral camptothecin gimatecan (ST1481) in patients with advanced solid tumors. Annal. Oncol. 2007, 18, 561–568. [Google Scholar] [CrossRef]
- Sergent, J.M.; Elgie, A.W.; Williamson, C.J.; Hill, B.T. Ex vivo effects of the dual topoisomerase inhibitor tafluposide (F 11782) on cells isolated from fresh tumor samples taken from patients with cancer. Anti-Cancer Drug 2003, 14, 467–473. [Google Scholar] [CrossRef]
- David-Cordonnier, M.H.; Laine, W.; Lansiaux, A.; Kouach, M.; Briand, G.; Pierré, A.; Hickman, J.A.; Bailly, C. Alkylation of Guanine in DNA by S23906-1, a Novel Potent Antitumor Compound Derived from the Plant Alkaloid Acronycine. Biochemistry 2002, 41, 9911–9920. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Sorba, G.; Pagliai, F.; Bussacca, S.; Genzzani, A.A. Medicinal Chemistry of Combretastatin A4: Present and Future Directions. J. Med. Chem. 2006, 49, 3033–3044. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Lippert, J.W.; Naraynan, V.R.; Varma, R.; Simpson, M.J.; Boyd, M.R.; Rener, G.A.; Bansal, N. Antineoplastic agents 322. synthesis of combretastatin A-4 prodrugs. Anti-Cancer Drug Des. 1995, 10, 299–309. [Google Scholar]
- Grossman, S.A.; Ye, X.; Peereboom, D.; Rosenfeld, M.R.; Mikkelsen, T.; Supko, J.G.; Desideri, S. Phase I study of terameprocol in patients with recurrent high-grade glioma. Neuro. Oncol. 2012, 14, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.G.; Feitelson, A.K.; Taylor, D.D. Inhibitory effect of genistein and daidzein on ovarian cancer cell growth. Anticancer Res. 2004, 24, 795–800. [Google Scholar]
- Mishra, S.; Karmodiya, K.; Surolia, N.; Surolia, A. Synthesis and exploration of novel curcumin analogues as anti-malarial agents. Bioorg. Med. Chem. 2008, 16, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Lal, J.; Gupta, S.K.; Thavaselvam, D.; Agrawal, D.D. Design, synthesis, synergistic antimicrobial activity and cytotoxicity of 4-aryl substituted 3,4-dihydropyrimidinones of curcumin. Bioorg. Med. Chem. 2012, 22, 2872–2876. [Google Scholar] [CrossRef]
- Sahu, P.K.; Sahu, P.K.; Gupta, S.K.; Thavaselvam, D.; Agarwal, D.D. Synthesis and evaluation of antimicrobial activity of 4H-pyrimido[2,1-b]benzothiazole, pyrazole and benzylidene derivatives of curcumin. Eur. J. Med. Chem. 2012, 54, 366–378. [Google Scholar] [CrossRef]
- Saja, K.; Babu, M.S.; Karunagaran, D.; Sudhakaran, P.R. Anti-inflammatory effect of curcumin involves down regulation of MMP-9 in blood mononuclear cells. Int. Immunopharm. 2007, 7, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Rai, D.; Yadav, D.; Bhargava, A.; Balzarini, J.; DeClercq, E. Synthesis, antibacterial and antiviral properties of curcumin bioconjugates bearing dipeptide, fatty acids and folic acid. Eur. J. Med. Chem. 2010, 45, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Zhichang, L.; Yinghong, W.; Yuanqin, Z.; Qinxiang, X. Synthesis and antibacterial activities of N-Substituted pyrazole curcumin derivatives. Chin. J. Org. Chem. 2012, 32, 1487–1492. [Google Scholar]
- Lee, W.H.; Loo, C.Y.; Bebawy, M.; Luk, F.; Mason, R.S.; Rohanizadeh, R. Curcumin and its Derivatives: Their Application in Neuropharmacology and Neuroscience in the 21st Century. Curr Neuropharmacol. 2013, 11, 338–378. [Google Scholar] [CrossRef] [PubMed]
- Yadav, I.S.; Nandekar, P.P.; Shrivastava, S.; Sanganwar, A.; Choudhry, A.; Agarwal, S.M. Ensemble docking and molecular dynamics identify knoevenagel curcumin derivatives with potent anti-EGFR activity. Gene 2014, 539, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Vyas, A.; Dandawate, P.; Padhye, S.; Ahmad, A.; Sarkar, F. Perspectives on new synthetic curcumin analogs and their potential anticancer properties. Curr. Pharm. Des. 2013, 19, 2047–2069. [Google Scholar] [PubMed]
- Balaji, S.N.; Ahsan, M.J.; Jadav, S.S.; Trivedi, V. Molecular modeling, synthesis and antimalarial potentials of curcumin analogues containing heterocyclic ring. Arab. J. Chem. 2019, 12, 2492–2500. [Google Scholar] [CrossRef]
- Rodrigues, F.C.; Kumar, N.V.A.; Thakur, G. The potency of heterocyclic curcumin analogues: An evidence-based review. Pharmacol. Res. 2012, 166, 105489. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.J.; Khalilullah, H.; Yasmin, S.; Jadav, S.S.; Govindasamy, J. Synthesis, characterisation, and in vitro anticancer activity of curcumin analogues bearing pyrazole/pyrimidine ring targeting EGFR tyrosine kinase. BioMed Res. Int. 2013, 2013, 239354. [Google Scholar] [CrossRef] [PubMed]
- Lhouvum, K.; Balaji, S.N.; Ahsan, M.J.; Trivedi, V. Plasmodium falciparum PFI1625c offers an opportunity to design potent anti-malarials: Biochemical characterization and testing potentials in drug discovery. Acta Trop. 2019, 191, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.J.; Sharma, S. Design, Synthesis and Anti-HIV Activity of Curcumin Analogues. In Lambert Academic Publishing; Lambert Academic Publishing: Saarbrücken, Germany, 2015; ISBN 978-3-659-75001-4. [Google Scholar]
- Sung, H.; Ferley, J.; Siegel, R.L.; Laversanne, M.; Soerjomartaram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Rayan, A.; Raiyn, J.; Falah, M. Nature is the best source of anticancer drugs: Indexing natural products for their anticancer bioactivity. PLoS ONE 2017, 12, e0187925. [Google Scholar] [CrossRef]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target Ther. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Weigman, V.J.; Fan, C.; Sawyer, L.R.; He, X.; Troester, M.A.; Sartor, C.I.; Rieger-House, T.; Bernard, P.S.; Carey, L.A.; et al. EGFR associated expression profiles vary with breast tumor subtype. BMC Genom. 2007, 8, 258. [Google Scholar] [CrossRef]
- Xu, H.; Yu, Y.; Marciniak, D.; Rishi, A.K.; Sarkar, F.H.; Kucuk, O.; Majumdar, A.P.N. Epidermal growth factor receptor (EGFR)–related protein inhibits multiple members of the EGFR family in colon and breast cancer cells. Mol. Cancer Ther. 2005, 4, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Rusnak, D.W.; Alligood, K.J.; Mullin, R.J.; Spehar, G.M.; Arenas-Elliott, C.; Martin, A.M.; Degenhardt, Y.; Rudolph, S.K.; Jr Haws, T.F.; Hudson-Curtis, B.L.; et al. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb®, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif. 2007, 40, 580–594. [Google Scholar] [CrossRef]
- Mohamady, S.; Galal, M.; Eldehna, W.M.; Gutierrez, D.C.; Ibrahim, H.S.; Elmazar, M.M.; Ali, H.I. Dual Targeting of VEGFR2 and C-Met Kinases via the Design and Synthesis of Substituted 3-(Triazolo-thiadiazin-3-yl)indolin-2-one Derivatives as Angiogenesis Inhibitors. ACS Omega 2020, 5, 18872–18886. [Google Scholar] [CrossRef] [PubMed]
- Sogabe, S.; Kawakita, Y.; Igaki, S.; Iwata, H.; Miki, H.; Cary, D.R.; Takagi, T.; Takagi, S.; Ohta, Y.; Ishikawa, T. Structure-Based Approach for the Discovery of Pyrrolo[3,2-d]pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors. ACS Med. Chem. Lett. 2013, 4, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Merla, A.; Goel, S. Novel Drugs Targeting the Epidermal Growth Factor Receptor and Its Downstream Pathways in the Treatment of Colorectal Cancer: A Systematic Review. Chemother. Res. Prac. 2012, 2012, 387172. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.J.; Johnson, D.E.; Grandis, J.R. EGFR-Targeted Therapies in the Post-Genomic Era. Cancer Metastasis Rev. 2017, 36, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.M.; Mitchell, M.S.; Mohan, R.S. Isolation of Curcumin from Turmeric. J. Chem. Edu. 2000, 77, 59–60. [Google Scholar] [CrossRef]
- Ahsan, M.J.; Meena, R.; Dubey, S.; Khan, V.; Manda, S.; Jadav, S.S.; Sharma, P.; Geesi, M.H.; Hassan, M.Z.; Bakht, M.A.; et al. Synthesis and biological potentials of some new 1,3,4-oxadiazole analogues. Med. Chem. Res. 2018, 27, 864–883. [Google Scholar] [CrossRef]
- Ahsan, M.J. Rationale Design, Synthesis And In Vitro Anticancer Activity of New 2,5-Disubstituted-1,3,4-Oxadiazole Analogues. ChemistrySelect 2016, 1, 4713–4720. [Google Scholar] [CrossRef]
- DTP Developmental Therapeutic Programs. Available online: http://dtp.nci.nih.gov (accessed on 19 August 2022).
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high flux anticancer drug screening using a diverse panel of cultured human tumor cell lines. J. Nat. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Boyd, M.R.; Paull, K.D. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Corona, P.; Carta, A.; Loriga, M.; Vitale, G.; Paglietti, G. Synthesis and in vitro antitumor activity of new quinoxaline derivatives. Eur. J. Med. Chem. 2009, 44, 1579–1591. [Google Scholar] [CrossRef]
- Grever, M.R.; Schepartz, S.A.; Chabner, B.A. The national cancer institute: Cancer drug discovery and development program. Sem. Oncol. 1992, 19, 622–638. [Google Scholar]
- Acton, E.M.; Narayanan, V.L.; Risbood, P.A.; Shoemaker, R.H.; Vistica, D.T.; Boyd, M.R. Anticancer Specificity of Some Ellipticinium Salts against Human Brain Tumors in vitro. J. Med. Chem. 1994, 37, 2185–2189. [Google Scholar] [CrossRef] [PubMed]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1998, 48, 589–601. [Google Scholar]
- Rostom, S.A.F. Synthesis and in vitro antitumor evaluation of some indeno[1,2-c]pyrazol(in)es substituted with sulfonamide, sulfonylurea(-thiourea) pharmacophores, and some derived thiazole ring systems. Bioorganic Med. Chem. 2006, 14, 6475–6485. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.A.; Rauh, D.; Kung, C.; Yun, C.H.; Fan, Q.; Rode, H.; Zhang, C.; Eck, M.J.; Weiss, W.A.; Shokat, K.M. Structure-guided development of affinity probes for tyrosine kinases using chemical genetics. Nat. Chem. Bio. 2007, 3, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.J.; Choudhary, K.; Jadav, S.S.; Yasmin, S.; Ansari, M.Y.; Sreenivasulsu, R. Synthesis, anticancer activity and molecular docking studies of curcumin analogues bearing pyrazole ring. Med. Chem. Res. 2015, 24, 4166–4180. [Google Scholar] [CrossRef]
- Sharma, R.; Singh, S.; Yasmin, S.; Bhatia, S.; Khalilullah, H.; Ahsan, M.J. Simple, efficient, and improved synthesis of Biginelli-type compounds of curcumin as anticancer agents. Med. Chem. Res. 2015, 24, 636–644. [Google Scholar] [CrossRef]
- X-ray Crystallographic Structure of EGFR. Available online: https://www.rcsb.org/structure/2j5f (accessed on 10 September 2022).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Panel | Cell Line | GP and %GI at 10 µM | C2 (NSC 781640) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1 (NSC 781637) | C2 (NSC 781640) | C3 (NSC 799008) | GI50 | Sub Panel MID b | Selectivity Ratio (MID a: MID b) | TGI | LC50 | |||||
| GP | %GI | GP | %GI | GP | %GI | |||||||
| Leukemia | CCRF-CEM | 44.18 | 55.82 | 10.44 | 89.66 | 55.57 | 44.43 | 3.21 | 3.06 | 0.97 | 70.6 | >100 |
| HL-60(TB) | 92.60 | 7.40 | 13.53 | 86.47 | 90.94 | 9.06 | 2.64 | 14.4 | >100 | |||
| K-562 | 72.65 | 17.35 | 9.21 | 90.79 | 74.68 | 25.32 | 3.11 | >100 | >100 | |||
| MOLT-4 | 42.97 | 57.03 | 9.71 | 90.29 | 52.53 | 47.47 | 3.38 | 22.6 | >100 | |||
| RPMI-8226 | 64.47 | 35.53 | 0.56 | 99.44 | 55.35 | 44.65 | 2.96 | 22.7 | >100 | |||
| SR | 36.42 | 63.58 | 4.12 | 95.88 | 67.80 | 32.20 | - | - | - | |||
| Non-Small Cell Lung Cancer | A549/ATCC | 98.11 | 1.89 | 44.15 | 55.85 | 78.13 | 21.87 | 4.03 | 3.29 | 0.91 | 15.0 | 52.6 |
| EKVX | 78.11 | 21.89 | 41.39 | 52.61 | 94.55 | 5.45 | 3.26 | 14.8 | 43.0 | |||
| HOP-62 | 90.11 | 9.89 | 50.58 | 49.42 | 99.14 | 0.86 | 3.41 | 12.8 | 37.8 | |||
| HOP-92 | 88.64 | 11.36 | 78.69 | 21.31 | - | - | 4.22 | 16.4 | 83.7 | |||
| NCI-H226 | 91.28 | 8.72 | 48.85 | 51.15 | 102.17 | −2.17 | 3.27 | 9.44 | 70.6 | |||
| NCI-H23 | 93.66 | 6.34 | 28.96 | 71.04 | 94.32 | 5.68 | 2.38 | 6.68 | 59.7 | |||
| NCI-H322M | 82.00 | 18.00 | 72.65 | 27.35 | 99.76 | 0.24 | 4.14 | 16.4 | 47.4 | |||
| NCI-H460 | 92.29 | 7.71 | 70.84 | 29.16 | 87.46 | 12.54 | 3.39 | 11.8 | 56.8 | |||
| NCI-H522 | 91.27 | 8.73 | 9.89 | 90.11 | 51.69 | 48.31 | 1.55 | 3.23 | 67.3 | |||
| Colon Cancer | COLO 205 | 91.32 | 8.68 | 71.81 | 28.19 | 114.89 | −14.89 | 2.84 | 2.19 | 1.35 | 8.95 | 40.5 |
| HCC-2998 | 103.85 | −3.85 | 41.77 | 58.23 | 103.02 | −3.02 | 1.83 | 3.53 | 6.82 | |||
| HCT-116 | 50.58 | 49.42 | 4.63 | 95.37 | 61.27 | 38.73 | 1.31 | 5.25 | 33.2 | |||
| HCT-15 | 85.38 | 14.62 | 10.28 | 89.72 | 92.27 | 7.73 | 2.44 | 9.66 | 48.4 | |||
| HT29 | −5.09 | 105.09 | −30.44 | 130.44 | 44.35 | 55.65 | 1.77 | 3.36 | 6.41 | |||
| KM12 | 76.99 | 23.01 | 12.12 | 87.88 | 86.85 | 13.15 | 2.02 | 5.28 | 28.7 | |||
| SW-620 | 85.58 | 14.42 | 10.43 | 89.57 | 101.39 | −1.39 | 3.11 | 1.05 | 54.1 | |||
| CNS Cancer | SF-268 | 76.05 | 23.95 | 19.85 | 80.15 | 84.32 | 15.68 | 3.12 | 2.82 | 1.05 | 13.7 | 60.2 |
| SF-295 | 97.33 | 2.67 | 75.37 | 25.63 | 104.21 | −4.21 | 2.87 | 9.52 | 32.6 | |||
| SF-539 | 78.08 | 21.92 | 3.35 | 96.65 | 97.11 | 2.89 | 2.18 | 6.92 | 29.0 | |||
| SNB-19 | 91.04 | 9.96 | 34.14 | 65.86 | 83.03 | 16.97 | 3.54 | 13.2 | 42.5 | |||
| SNB-75 | 69.72 | 30.28 | 49.42 | 51.58 | 80.29 | 9.71 | - | - | - | |||
| U251 | 75.53 | 24.47 | 8.54 | 91.46 | 72.05 | 27.95 | 2.37 | 6.36 | −22.5 | |||
| Melanoma | LOX IMVI | 61.57 | 38.43 | 2.83 | 97.17 | 78.03 | 21.97 | 1.68 | 2.63 | 1.13 | 3.57 | 7.56 |
| MALME-3M | 99.96 | 0.04 | 58.79 | 41.21 | 91.82 | 8.18 | 3.88 | 15.7 | 42.2 | |||
| M14 | 88.30 | 11.70 | 7.71 | 42.29 | 92.06 | 7.94 | 2.38 | 6.87 | 27.5 | |||
| MDA-MB-435 | 74.93 | 25.07 | 4.57 | 95.43 | 94.71 | 5.29 | 1.63 | 5.98 | 25.3 | |||
| SK-MEL-2 | 92.56 | 7.44 | 82.92 | 17.08 | 83.56 | 16.44 | 3.30 | 12. | 39.2 | |||
| SK-MEL-28 | 81.58 | 18.42 | 17.59 | 82.41 | 98.97 | 1.03 | 2.74 | 10.1 | 40.5 | |||
| SK-MEL-5 | - | - | - | - | 83.18 | 16.82 | 2.18 | 4.86 | 12.3 | |||
| UACC-257 | 96.99 | 3.01 | 41.10 | 58.9 | 67.98 | 32.02 | 3.32 | 11.3 | 33.7 | |||
| UACC-62 | 80.58 | 9.42 | 38.17 | 61.83 | 75.14 | 24.86 | 2.59 | 7.42 | 29.3 | |||
| Ovarian Cancer | IGROV1 | 8.57 | 91.43 | 33.46 | 66.54 | 99.85 | 0.15 | 3.01 | 3.32 | 0.89 | 12.3 | 58.5 |
| OVCAR-3 | 83.75 | 16.25 | 10.66 | 89.34 | 93.60 | 6.40 | 3.19 | 12.1 | >100 | |||
| OVCAR-4 | 78.34 | 21.66 | 56.26 | 43.74 | 100.98 | −0.98 | 4.61 | 16.9 | 41.1 | |||
| OVCAR-5 | 103.44 | −3.44 | 57.58 | 42.42 | 114.83 | −14.83 | 2.76 | 11.0 | 37.9 | |||
| OVCAR-8 | 107.34 | −7.34 | 7.53 | 92.47 | 82.79 | 17.21 | 3.34 | 10.5 | 50.3 | |||
| NCI/ADR-RES | 96.51 | 3.49 | 55.21 | 44.79 | 99.97 | 0.03 | 3.53 | 22.0 | >100 | |||
| SK-OV-3 | 89.73 | 9.27 | 47.71 | 52.29 | 102.00 | −2.00 | 2.78 | 10.6 | 35.1 | |||
| Renal Cancer | 786-0 | 92.56 | 7.44 | 29.70 | 70.30 | 95.36 | 4.64 | 2.74 | 2.93 | 1.01 | 8.51 | 31.0 |
| A498 | 102.35 | −2.35 | 78.54 | 21.46 | 97.96 | 2.04 | 3.79 | 13.2 | 36.4 | |||
| ACHN | 83.10 | 16.90 | 37.78 | 62.22 | 93.04 | 6.96 | 2.84 | 12.7 | 66.8 | |||
| CAKI-1 | 92.92 | 7.08 | 63.18 | 36.82 | 83.20 | 16.80 | 1.46 | 30.0 | 62.0 | |||
| RXF 393 | 90.70 | 9.30 | 11.82 | 88.18 | 106.20 | −6.20 | 2.18 | 4.92 | >100 | |||
| SN 12C | 80.80 | 9.20 | 27.84 | 72.16 | 76.04 | 23.96 | 4.82 | 19.5 | 65.7 | |||
| TK-10 | 117.99 | −17.99 | 86.02 | 13.98 | 102.66 | −2.66 | 2.91 | 10.1 | 36.1 | |||
| UO-31 | 64.88 | 35.12 | 25.16 | 74.86 | 72.46 | 27.54 | 2.74 | 12.3 | 36.6 | |||
| Prostate Cancer | PC-3 | 77.28 | 27.72 | 34.86 | 65.14 | 82.94 | 17.06 | 4.68 | 4.08 | 0.73 | 25.3 | >100 |
| DU-145 | 91.71 | 8.29 | 28.74 | 71.26 | 84.61 | 16.39 | 3.48 | 13.0 | 37.0 | |||
| Breast Cancer | MCF7 | 47.62 | 52.38 | 13.75 | 86.25 | 67.34 | 32.66 | 2.00 | 3.23 | 0.92 | 11.0 | 78.2 |
| MDA-MB-231 | 60.31 | 39.69 | 29.00 | 71.00 | 65.84 | 34.16 | 3.64 | 15.1 | >100 | |||
| HS 578T | 87.44 | 12.66 | 36.31 | 63.69 | 85.47 | 14.53 | 5.48 | >100 | >100 | |||
| BT-549 | 66.90 | 33.10 | 15.17 | 84.83 | 80.77 | 9.23 | 2.07 | 66.7 | 28.6 | |||
| T-47D | 85.96 | 14.04 | 24.11 | 75.89 | 82.52 | 7.48 | 3.44 | 22.7 | >100 | |||
| MDA-MB-468 | 76.44 | 23.56 | 29.59 | 70.41 | 89.61 | 10.39 | 2.76 | 7.19 | 44.5 | |||
| Mean | 79.63 | 20.37 | 31.78 | 68.22 | 85.67 | 14.33 | 2.97 | |||||
| Range (% GI) | −17.99 to 105.09 | 13.98 to 130.44 | −14.83 to 55.65 | |||||||||
| Total number of cell lines and sum of concentration (µM) | 59 | 58/172.3 | ||||||||||
| MID a | 2.971 | |||||||||||
| Panel | C1 | C2 | C3 | Cur * | Gefitinib # | Imatinib # |
|---|---|---|---|---|---|---|
| Leukemia | 39.45 | 92.09 | 33.86 | 97.76 | 79.68 | 9 |
| Non-Small cell lung cancer | 10.50 | 49.78 | 11.60 | 49.27 | 63.97 | 15.68 |
| Colon Cancer | 30.20 | 82.77 | 13.71 | 95.76 | 52.19 | 5.34 |
| CNS Cancer | 18.88 | 68.56 | 11.50 | 60.75 | 46.13 | 5.8 |
| Melanoma | 14.19 | 62.04 | 14.95 | 54.63 | 44.99 | −0.87 |
| Ovarian Cancer | 18.76 | 61.66 | 0.85 | 44.66 | 60.93 | −7.16 |
| Renal Cancer | 8.09 | 54.99 | 9.14 | 45.35 | 77.89 | 3.25 |
| Prostate Cancer | 18.01 | 68.20 | 16.73 | 61.3 | 59.6 | 12.5 |
| Breast Cancer | 29.24 | 75.35 | 18.08 | 56.1 | 52.88 | 12.15 |
| S. No. | Compound | 2D Docking | Docking Score | Glide Emodel | Interaction |
|---|---|---|---|---|---|
| 1 | C1 |  | −5.117 | −66.925 | Aromatic H-Bond (Asp855) |
| 2 | C2 |  | −5.086 | −62.292 | H-Bond (Thr790 and Thr854); π-Cationic (Arg841); Aromatic H-Bond (Asn842) |
| 3 | C3 |  | −4.396 | −66.538 | H-Bond (Thr790, Thr854, Lys875); Halogen Bond (Lys875); Aromatic H-Bond (Asp855) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afzal, O.; Yusuf, M.; Ahsan, M.J.; Altamimi, A.S.A.; Bakht, M.A.; Ali, A.; Salahuddin. Chemical Modification of Curcumin into Its Semi-Synthetic Analogs Bearing Pyrimidinone Moiety as Anticancer Agents. Plants 2022, 11, 2737. https://doi.org/10.3390/plants11202737
Afzal O, Yusuf M, Ahsan MJ, Altamimi ASA, Bakht MA, Ali A, Salahuddin. Chemical Modification of Curcumin into Its Semi-Synthetic Analogs Bearing Pyrimidinone Moiety as Anticancer Agents. Plants. 2022; 11(20):2737. https://doi.org/10.3390/plants11202737
Chicago/Turabian StyleAfzal, Obaid, Mohammad Yusuf, Mohamed Jawed Ahsan, Abdulmalik S. A. Altamimi, Md. Afroz Bakht, Amena Ali, and Salahuddin. 2022. "Chemical Modification of Curcumin into Its Semi-Synthetic Analogs Bearing Pyrimidinone Moiety as Anticancer Agents" Plants 11, no. 20: 2737. https://doi.org/10.3390/plants11202737
APA StyleAfzal, O., Yusuf, M., Ahsan, M. J., Altamimi, A. S. A., Bakht, M. A., Ali, A., & Salahuddin. (2022). Chemical Modification of Curcumin into Its Semi-Synthetic Analogs Bearing Pyrimidinone Moiety as Anticancer Agents. Plants, 11(20), 2737. https://doi.org/10.3390/plants11202737