
Molecular Identification of African Nymphaea Species (Water Lily) Based on ITS, trnT-trnF and rpl16

 , ,
, , 
Abstract
1. Introduction
2. Results
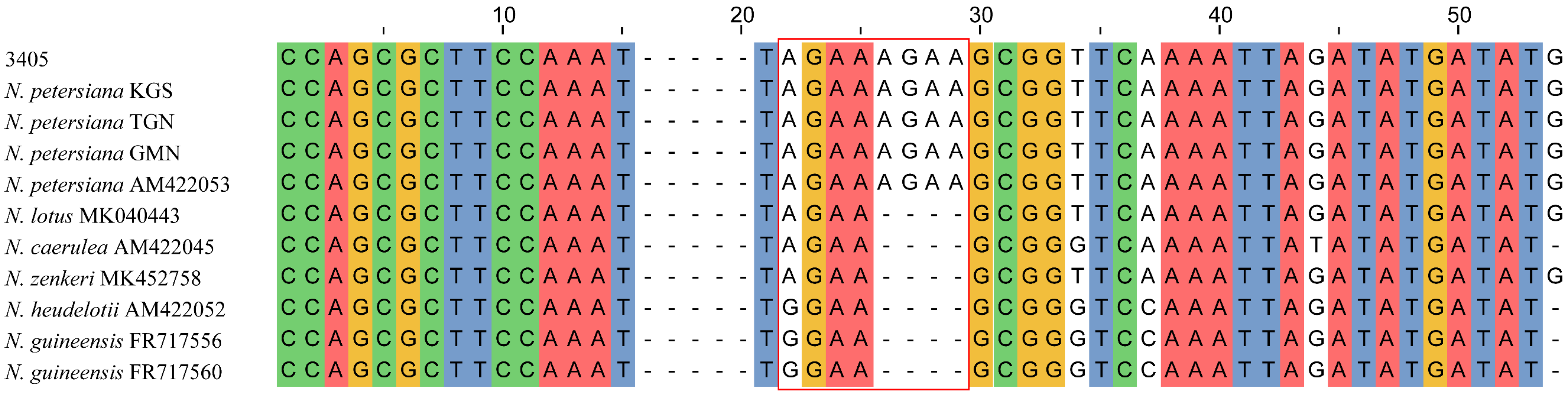
2.1. DNA Barcodes Universality and Sequence Characteristics
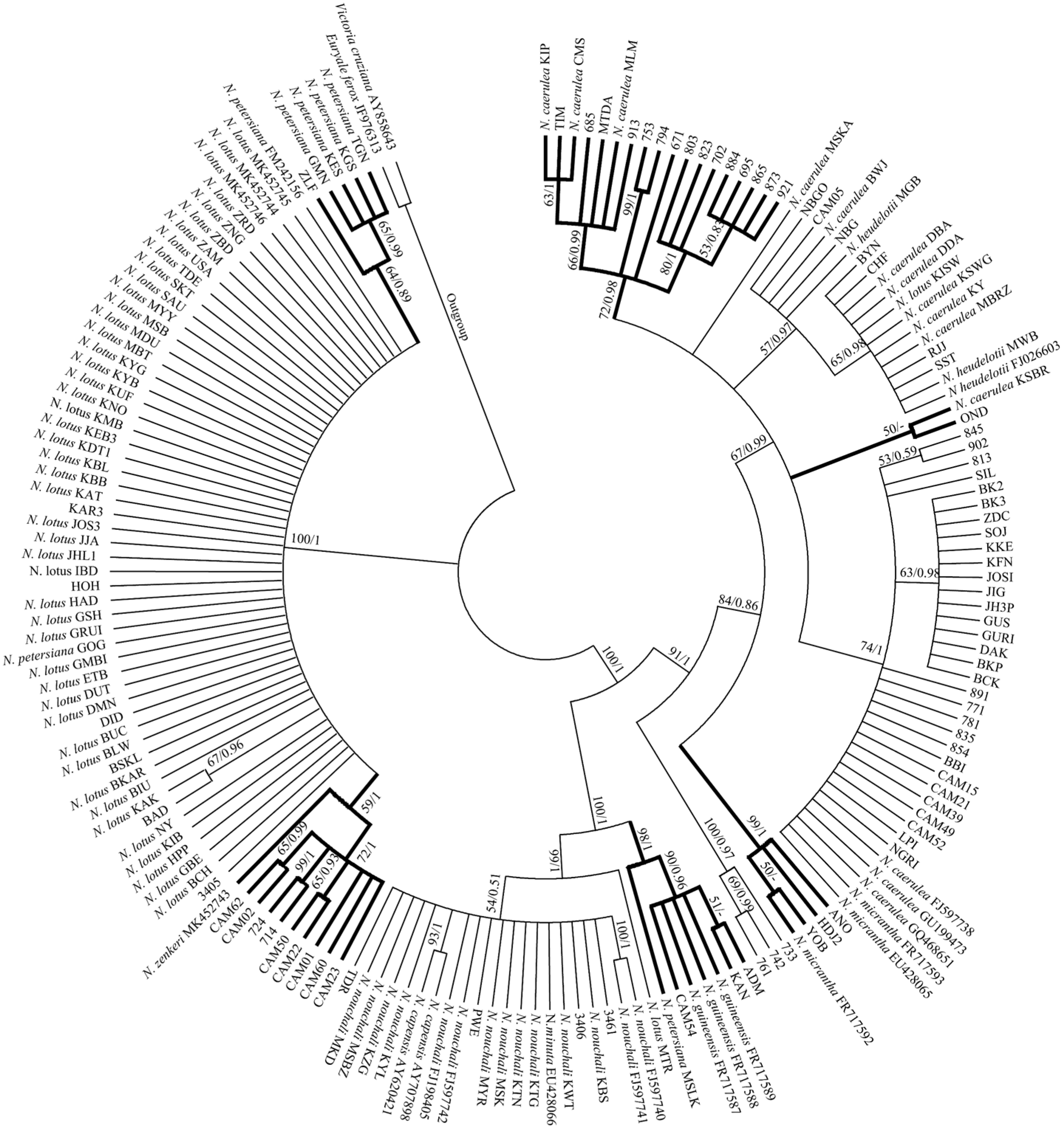
2.2. Species Identification of African Nymphaea Species Based on DNA Barcoding Analysis
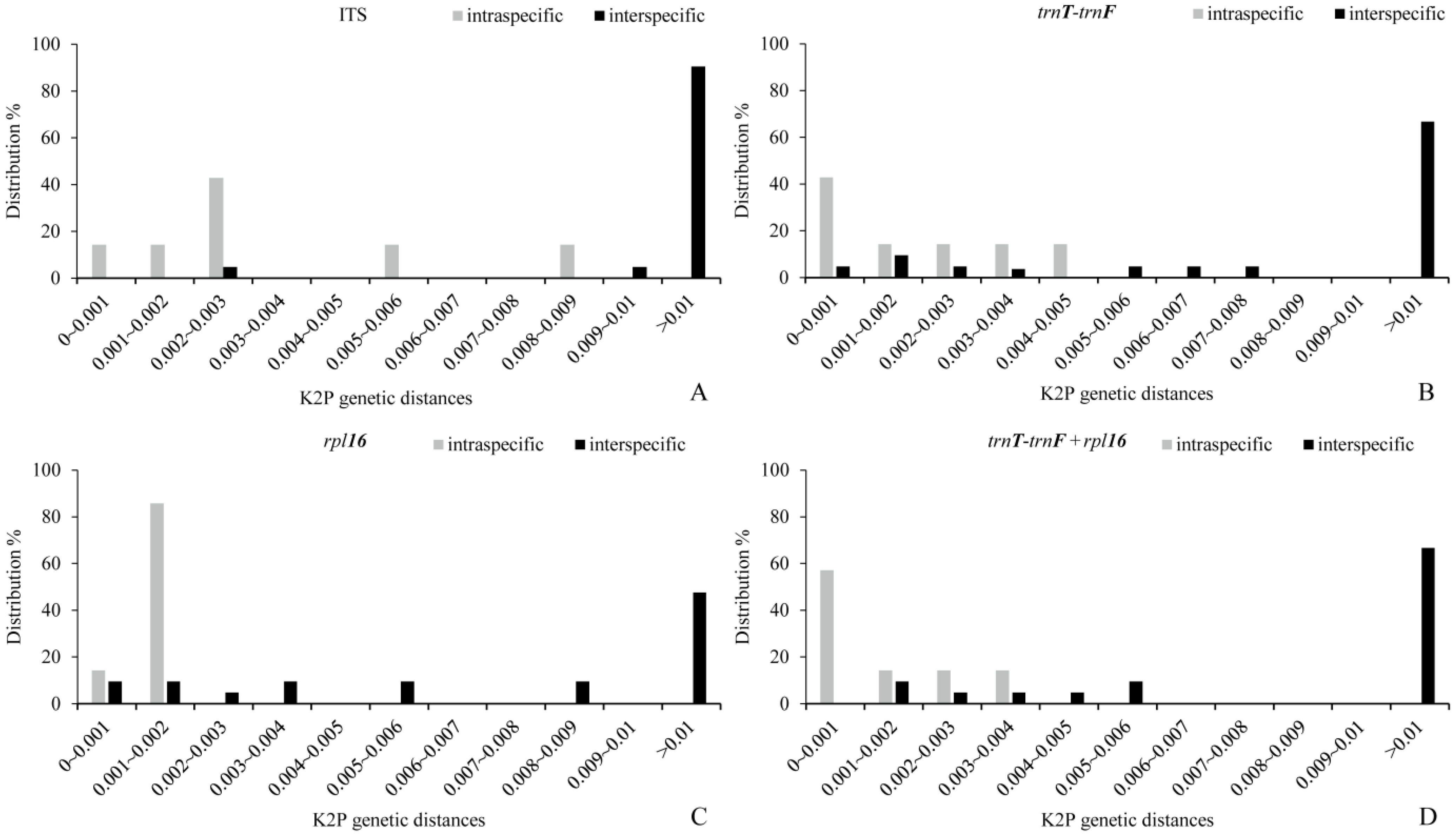
2.3. Genetic Differences and Barcoding Gap Analysis of Candidate DNA Barcoding
3. Discussion
4. Materials and Methods
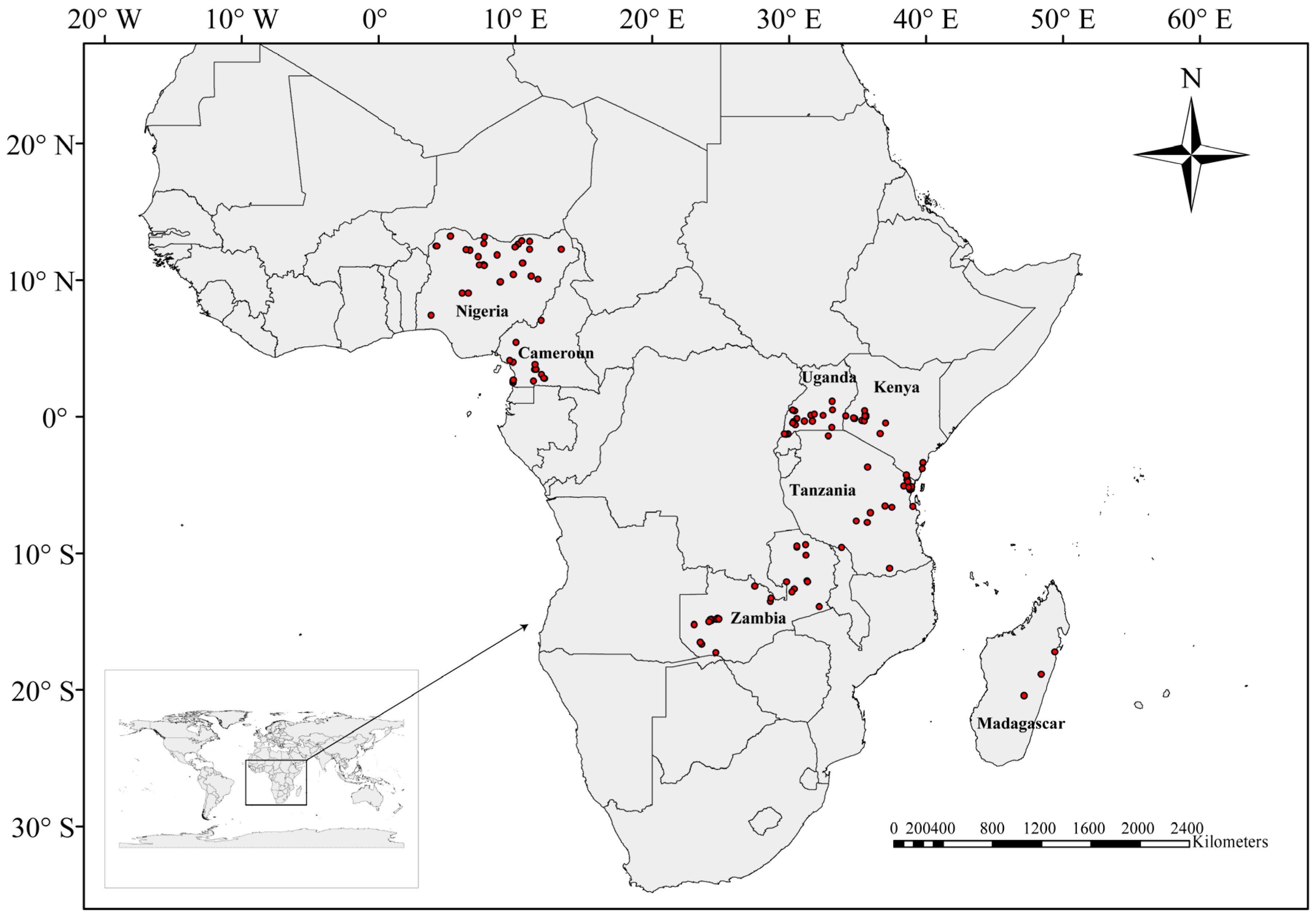
4.1. Sample Collection
4.2. DNA Isolation, PCR Amplification and Sequencing
4.3. Sequence Alignment and Data Analysis
4.4. Genetic Divergence Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schneider, E.L.; Williamson, P.S. Nymphaeaceae. In Families and Genera of Flowering Plants; Kubitzki, K., Ed.; Springer: New York, NY, USA, 1993; Volume 2, pp. 486–493. [Google Scholar]
- Borsch, T.; Hilu, K.W.; Wiersema, J.H.; Löhne, C.; Barthlott, W.; Wilde, V. Phylogeny of Nymphaea (Nymphaeaceae): Evidence from substitutions and microstructural changes in the chloroplast trnT-trnF region. Int. J. Mol. Sci. 2007, 168, 639–671. [Google Scholar] [CrossRef]
- Biswal, D.K.; Debnath, M.; Kumar, S.; Tandon, P. Phylogenetic reconstruction in the Order Nymphaeales: ITS2 secondary structure analysis and in silico testing of maturase k (matK) as a potential marker for DNA barcoding. BMC Bioinform. 2012, 13, S26. [Google Scholar] [CrossRef] [PubMed]
- Gruenstaeudl, M.; Nauheimer, L.; Borsch, T. Plastid genome structure and phylogenomics of Nymphaeales: Conserved gene order and new insights into relationships. Plant Syst. Evol. 2017, 303, 1251–1270. [Google Scholar] [CrossRef]
- Conard, H.S. The Waterlilies: A Monograph of the Genus Nymphaea; Publications of the Carnegie Institutions of Washington: Washington, DC, USA, 1905; Volume 4, pp. 1–279. [Google Scholar]
- Dkhar, J.; Kumaria, S.; Rao, S.R.; Tandon, P. Sequence characteristics and phylogenetic implications of the nrDNA internal transcribed spacers (ITS) in the genus Nymphaea with focus on some Indian representatives. Plant Syst. Evol. 2012, 298, 93–108. [Google Scholar] [CrossRef]
- Borsch, T.; Löhne, C.; Mbaye, M.S.; Wiersema, J.H. Towards a complete species level tree of Nymphaea: Shedding further light on subg. Brachyceras and its relationships to the Australian waterlilies. Telopea 2011, 13, 193–217. [Google Scholar] [CrossRef]
- Guruge, S.; Yakandawala, D.; Yakandawala, K. A taxonomic synopsis of Nymphaea nouchali Burm. f. and infraspecific taxa. J. Natl. Sci. Found Sri 2017, 45, 307–318. [Google Scholar] [CrossRef]
- Borsch, T.; Wiersema, J.H.; Hellquist, C.B.; Löhne, C.; Govers, K. Speciation in North American water lilies: Evidence for the hybrid origin of the newly discovered Canadian endemic Nymphaea loriana sp. nov. (Nymphaeaceae) in a past contact zone. Botany-Botanique 2014, 92, 867–882. [Google Scholar] [CrossRef]
- Baldwin, B.G.; Sanderson, M.J.; Porter, J.M.; Wojciechowski, M.F.; Donoghue, C.M.J. The ITS region of nuclear ribosomal DNA: A valuable source of evidence on angiosperm phylogeny. Ann. Mo. Bot. Gard. 1995, 82, 247–277. [Google Scholar] [CrossRef]
- Soltis, D.; Soltis, P.; Chase, M.; Mort, M.; Albach, D.; Zanis, M.; Savolainen, V.; Hahn, W.; Hoot, S.; Fay, M. Angiosperm phylogeny inferred from a combined data set of 18S rDNA, rbcL, and atpB sequences. Bot. J. Linn. Soc. 2000, 133, 381–461. [Google Scholar] [CrossRef]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef] [PubMed]
- Kress, W.J.; Wurdack, K.J.; Zimmer, E.A.; Weigt, L.A.; Janzen, D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 2005, 102, 8369–8374. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, P.M.; Forrest, L.L.; Spouge, J.L.; Hajibabaei, M.; Little, D.P. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar] [CrossRef]
- Karapatzak, E.; Krigas, N.; Ganopoulos, I.; Papanastasi, K.; Kyrkas, D.; Yfanti, P.; Nikisianis, N.; Karydas, A.; Manthos, I.; Kosma, I.S.; et al. Documenting greek indigenous germplasm of cornelian cherry (Cornus mas L.) for sustainable utilization: Molecular authentication, asexual propagation, and phytochemical evaluation. Plants 2022, 11, 1345. [Google Scholar] [CrossRef]
- Wang, J.; Fu, C.N.; Mo, Z.Q.; Möller, M.; Yang, J.B.; Zhang, Z.R.; Li, D.Z.; Gao, L.M. Testing the complete plastome for species discrimination, cryptic species discovery and phylogenetic resolution in Cephalotaxus (Cephalotaxaceae). Front. Plant Sci. 2022, 13, 768810. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, P.M.; Graham, S.W.; Little, D.P. Choosing and using a plant DNA barcode. PLoS ONE 2011, 6, e19254. [Google Scholar] [CrossRef]
- Chase, M.W.; Cowan, R.S.; Hollingsworth, P.M.; van den Berg, C.; Madriñán, S.; Petersen, G.; Seberg, O.; Jørgsensen, T.; Cameron, K.M.; Carine, M.; et al. A proposal for a standardised protocol to barcode all land plants. Taxon 2007, 56, 295–299. [Google Scholar] [CrossRef]
- Kress, W.J.; Erickson, D.L. A two-locus global DNA barcode for land plants: The coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS ONE 2007, 2, e508. [Google Scholar] [CrossRef]
- Hollingsworth, M.L.; Andra, C.A.; Forrest, L.L.; Richardson, J.; Pennington, R.T.; Long, D.G.; Cowan, R.; Chase, M.W.; Gaudeul, M.; Hollingsworth, P.M. Selecting barcoding loci for plants: Evaluation of seven candidate loci with species-level sampling in three divergent groups of land plants. Mol. Ecol. Resour. 2009, 9, 439–457. [Google Scholar] [CrossRef]
- Löhne, C.; Borsch, T.; Jacobs, S.W.L.; Hellquist, C.B.; Wiersema, J.H. Nuclear and plastid DNA sequences reveal complex reticulate patterns in Australian water-lilies (Nymphaea subgenus Anecphya, Nymphaeaceae). Aust. Syst. Bot. 2008, 21, 229–250. [Google Scholar] [CrossRef]
- Puillandre, N.; Brouillet, S.; Achaz, G. ASAP: Assemble species by automatic partitioning. Mol. Ecol. Resour. 2021, 21, 609–620. [Google Scholar] [CrossRef]
- Meyer, C.P.; Paulay, G. DNA barcoding: Error rates based on comprehensive sampling. PLoS Biol. 2005, 3, e422. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Gielly, L.; Pautou, G.; Bouvet, J. Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol. Biol. 1991, 17, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Naugžemys, D.; Patamsytė, J.; Žilinskaitė, S.; Hoshino, Y.; Skridaila, A.; Žvingila, D. Genetic structure of native blue honeysuckle populations in the western and eastern Eurasian ranges. Plants 2022, 11, 1480. [Google Scholar] [CrossRef] [PubMed]
- Borsch, T.; Hilu, K.W.; Quandt, D.; Wilde, V.; Neinhuis, C.; Barthlott, W. Noncoding plastid trnT-trnF sequences reveal a well resolved phylogeny of basal angiosperms. J. Evol. Biol. 2003, 16, 558–576. [Google Scholar] [CrossRef]
- Borsch, T.; Lohne, C.; Wiersema, J. Phylogeny and evolutionary patterns in Nymphaeales: Integrating genes, genomes and morphology. Taxon 2008, 57, 1052–1081. [Google Scholar] [CrossRef]
- China Plant BOL Group. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. USA 2011, 108, 19641–19646. [Google Scholar] [CrossRef]
- Mao, K.; Hao, G.; Liu, J.; Adams, R.P.; Milne, R.I. Diversification and biogeography of Juniperus (Cupressaceae): Variable diversification rates and multiple intercontinental dispersals. New Phytol. 2010, 188, 254–272. [Google Scholar] [CrossRef]
- Li, Z.Z.; Ngarega, B.K.; Lehtonen, S.; Gichira, A.W.; Karichu, M.J.; Wang, Q.F.; Chen, J.M. Cryptic diversity within the African aquatic plant Ottelia ulvifolia (Hydrocharitaceae) revealed by population genetic and phylogenetic analyses. J. Plant Res. 2020, 133, 373–381. [Google Scholar] [CrossRef]
- Zhang, G.Q.; Chen, G.Z.; Chen, L.J.; Zhai, J.W.; Huang, J.; Wu, X.Y.; Li, M.H.; Peng, D.H.; Rao, W.H.; Liu, Z.J.; et al. Phylogenetic incongruence in Cymbidium orchids. Plant Divers. 2021, 43, 452–461. [Google Scholar] [CrossRef]
- Hollingsworth, P.M.; Li, D.Z.; Michelle, V.D.B.; Twyford, A.D. Telling plant species apart with DNA: From barcodes to genomes. Philos. Trans. R. Soc. B. 2016, 371, 20150338. [Google Scholar] [CrossRef]
- Fu, C.N.; Wu, C.S.; Ye, L.J.; Mo, Z.Q.; Liu, J.; Chang, Y.W.; Li, D.Z.; Chaw, S.M.; Gao, L.M. Prevalence of isomeric plastomes and effectiveness of plastome super-barcodes in yews (Taxus) worldwide. Sci. Rep. 2019, 9, 2773. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.H.; Liu, C.K.; Yang, Z.Y.; Yang, L.F.; He, Z.S.; Wang, H.C.; Yang, J.B.; Yi, T.S. Testing and using complete plastomes and ribosomal DNA sequences as the next generation DNA barcodes in Panax (Araliaceae). Mol. Ecol. Resour. 2019, 19, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.N.; Mo, Z.Q.; Yang, J.B.; Cai, J.; Ye, L.J.; Zou, J.Y.; Qin, H.T.; Zheng, W.; Hollingsworth, P.M.; Li, D.Z.; et al. Testing genome skimming for species discrimination in the large and taxonomically difficult genus Rhododendron. Mol. Ecol. Resour. 2022, 22, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Sauquet, H.; Doyle, J.A.; Scharaschkin, T.; Borsch, T.; Hilu, K.W.; Chatrou, L.W.; Thomas, A.L. Phylogenetic analysis of Magnoliales and Myristicaceae based on multiple datasets: Implications for character evolution. Bot. J. Linn. Soc. 2003, 142, 125–186. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for big datasets. Mol. Biol. Evol 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, M.; Dong, X.Y.; Lin, R.Z.; Fan, J.H.; Chen, Z.D. Evaluation of four commonly used DNA barcoding Loci for Chinese medicinal plants of the family Schisandraceae. PLoS ONE 2015, 10, e0125574. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequences | Percentage PCR Success (%) | Percentage Sequencing Success (%) | Aligned Sequence Length (bp) | GC Content (%) | Variable Sites | Percentage Variable (%) | Parsimony Informative Sites |
|---|---|---|---|---|---|---|---|
| ITS | 100 | 99.37 | 731 | 51.8 | 212 | 29.73 | 196 |
| trnT-trnF | 98.1 | 96.2 | 1507 | 36.61 | 126 | 8.36 | 70 |
| rpl16 | 100 | 100 | 766 | 36.1 | 34 | 4.44 | 15 |
| Sequences | No. of Samples | The Range of Interspecific Distance | The Range of Intraspecific Distance | Average Interspecific Distance | Average Intraspecific Distance |
|---|---|---|---|---|---|
| ITS | 144 | 0.002–0.261 | 0–0.009 | 0.159 ± 0.016 | 0.004 ± 0.001 |
| trnT-trnF | 141 | 0.002–0.033 | 0.001–0.004 | 0.02 ± 0.003 | 0.002 ± 0.0006 |
| rpl16 | 144 | 0–0.014 | 0–0.001 | 0.008 ± 0.003 | 0.0006 ± 0.0003 |
| trnT-trnF+rpl16 | 141 | 0.001–0.027 | 0.001–0.003 | 0.018 ± 0.003 | 0.0015 ± 0.0004 |
| W+ | W− | RELATIVE Ranks | N | p-Value≤ | Result | |
|---|---|---|---|---|---|---|
| W+ | W− | |||||
| ITS | trnT-trnF | 206 | 5 | 21 | 0.001 | ITS > trnT-trnF |
| ITS | rpl16 | 206 | 4 | 21 | 0.001 | ITS > rpl16 |
| ITS | trnT-trnF+rpl16 | 226 | 5 | 21 | 0.001 | ITS > trnT-trnF+rpl16 |
| trnT-trnF | rpl16 | 203.5 | 27.5 | 21 | 0.003 | trnT-trnF > rpl16 |
| trnT-trnF | trnT-trnF+rpl16 | 187 | 3 | 21 | 0.001 | trnT-trnF > trnT-trnF+rpl16 |
| rpl16 | trnT-trnF+rpl16 | 28.5 | 202.5 | 21 | 0.003 | rpl16 < trnT-trnF+rpl16 |
| ITS | trnT-trnF | 206 | 5 | 21 | 0.001 | ITS > trnT-trnF |
| ITS | rpl16 | 206 | 4 | 21 | 0.001 | ITS > rpl16 |
| ITS | trnT-trnF+rpl16 | 226 | 5 | 21 | 0.001 | ITS > trnT-trnF+rpl16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, Z.-H.; Munywoki, J.M.; Wang, Q.-F.; Malombe, I.; Li, Z.-Z.; Chen, J.-M. Molecular Identification of African Nymphaea Species (Water Lily) Based on ITS, trnT-trnF and rpl16. Plants 2022, 11, 2431. https://doi.org/10.3390/plants11182431
Qian Z-H, Munywoki JM, Wang Q-F, Malombe I, Li Z-Z, Chen J-M. Molecular Identification of African Nymphaea Species (Water Lily) Based on ITS, trnT-trnF and rpl16. Plants. 2022; 11(18):2431. https://doi.org/10.3390/plants11182431
Chicago/Turabian StyleQian, Zhi-Hao, Justus Mulinge Munywoki, Qing-Feng Wang, Itambo Malombe, Zhi-Zhong Li, and Jin-Ming Chen. 2022. "Molecular Identification of African Nymphaea Species (Water Lily) Based on ITS, trnT-trnF and rpl16" Plants 11, no. 18: 2431. https://doi.org/10.3390/plants11182431
APA StyleQian, Z.-H., Munywoki, J. M., Wang, Q.-F., Malombe, I., Li, Z.-Z., & Chen, J.-M. (2022). Molecular Identification of African Nymphaea Species (Water Lily) Based on ITS, trnT-trnF and rpl16. Plants, 11(18), 2431. https://doi.org/10.3390/plants11182431