
Genome-Wide Identification and Spatial Expression Analysis of Histone Modification Gene Families in the Rubber Dandelion Taraxacum kok-saghyz

 ,
,  ,
,  , , , ,
, , , , 
Abstract
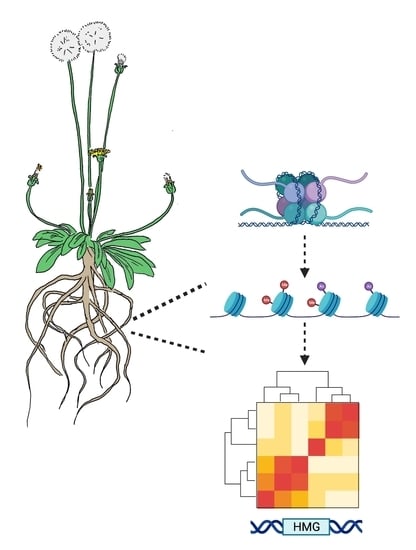
:
1. Introduction
2. Results
2.1. HMG Genes Identification
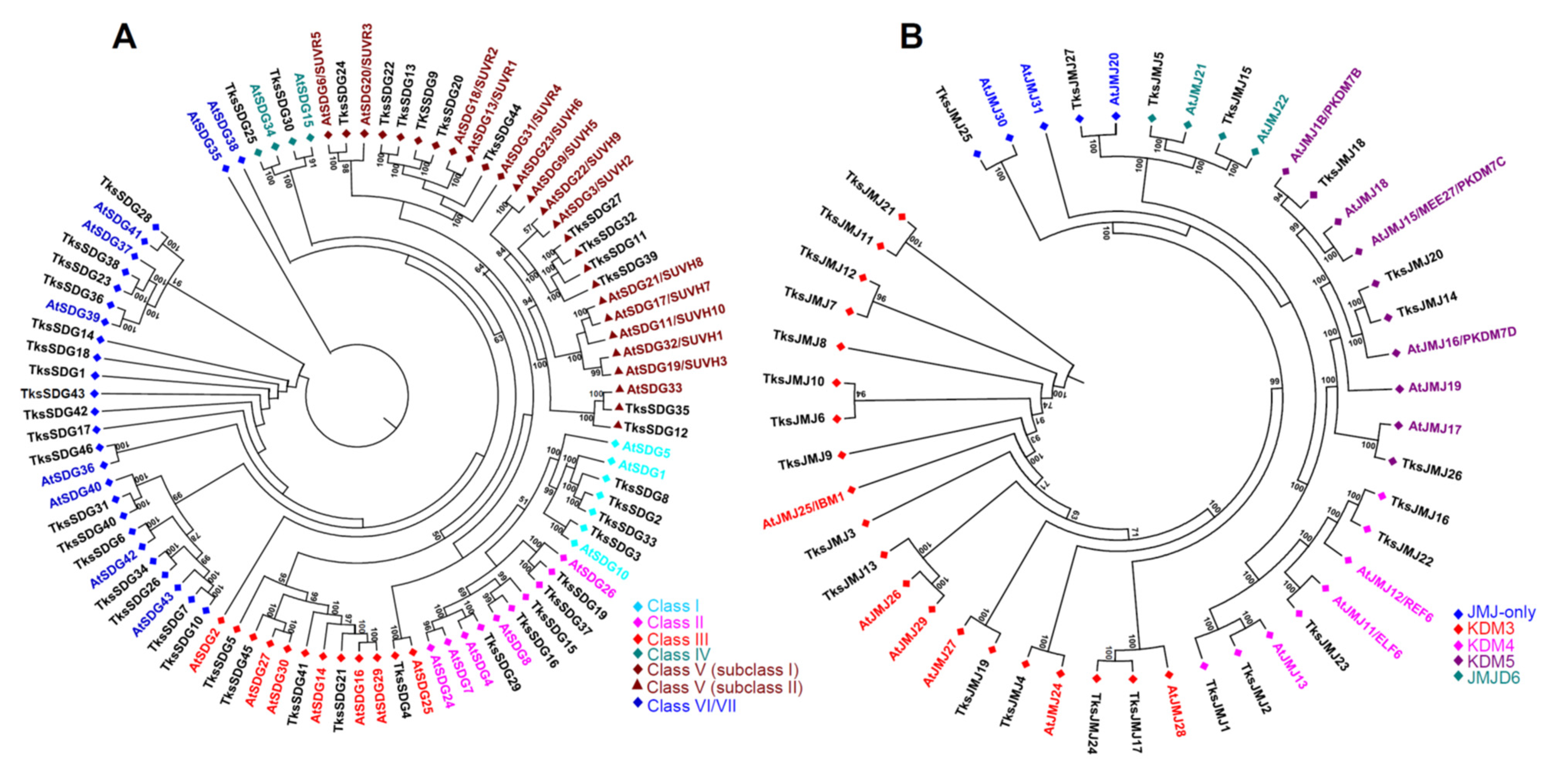
2.1.1. HMTs
2.1.2. HDMs
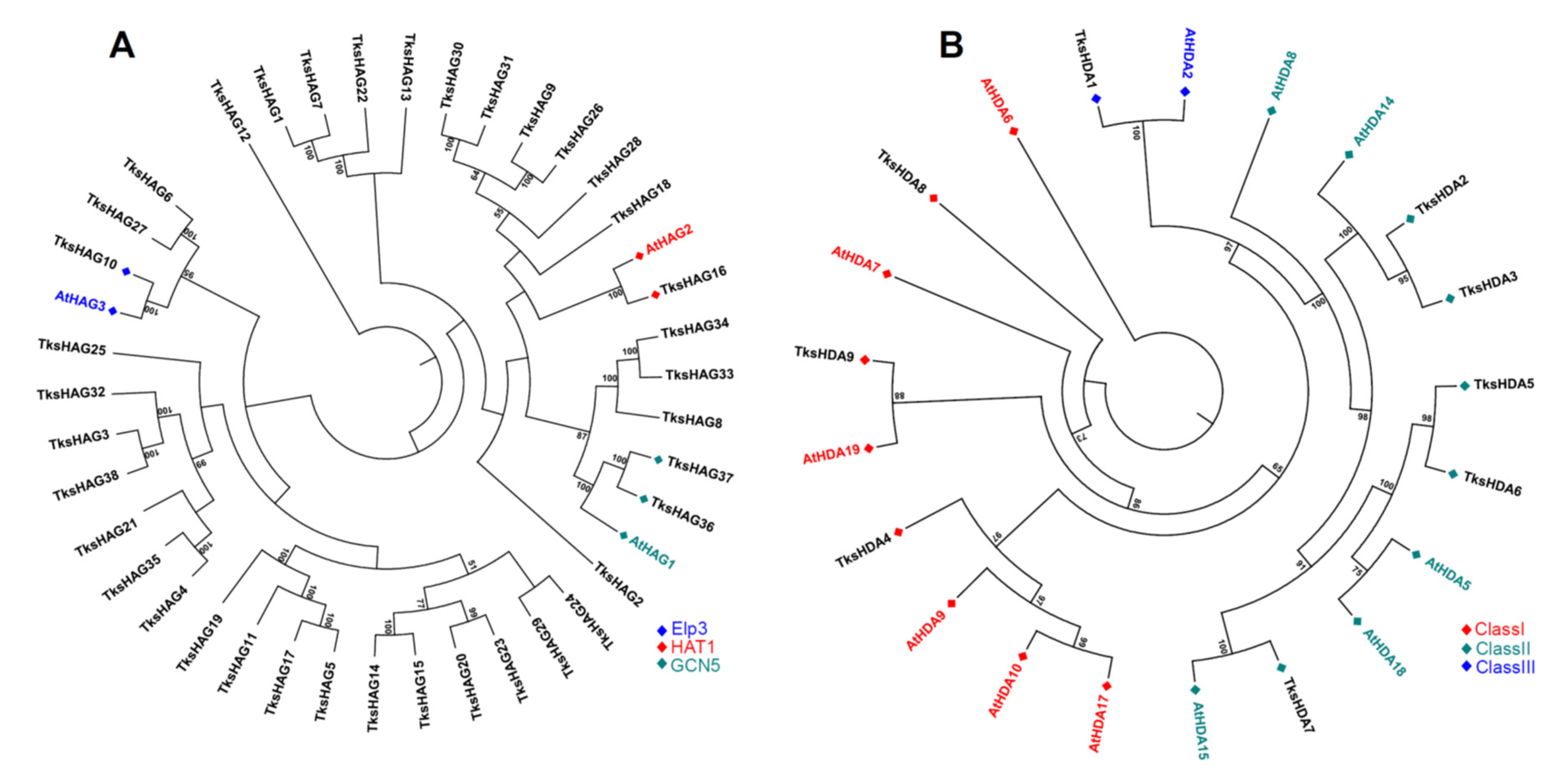
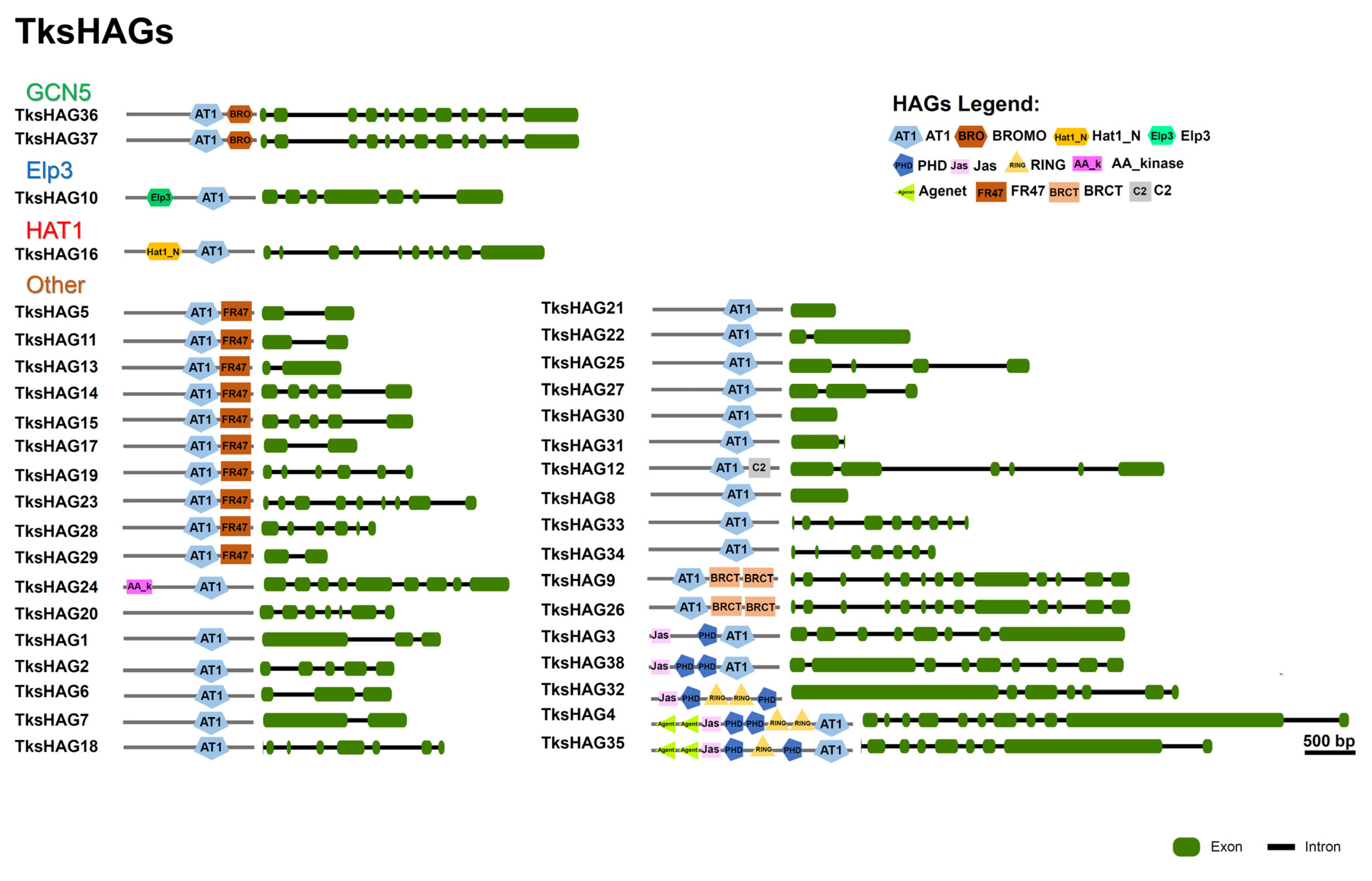
2.1.3. HATs
2.1.4. HDACs
2.2. In Silico Expression Analysis of HMG Genes in Different Organs and Developmental Stages
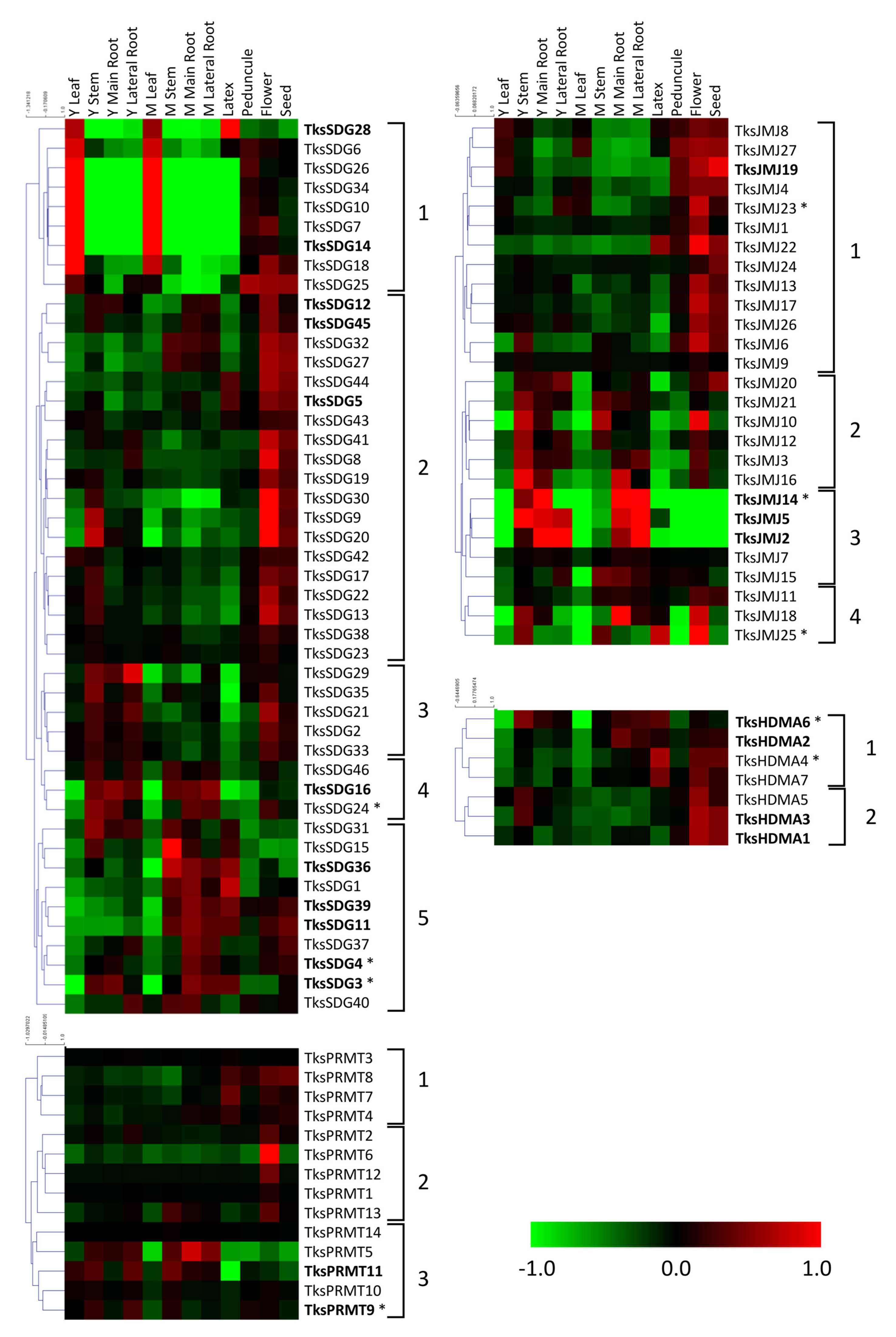
2.2.1. HMTs
2.2.2. HDMs
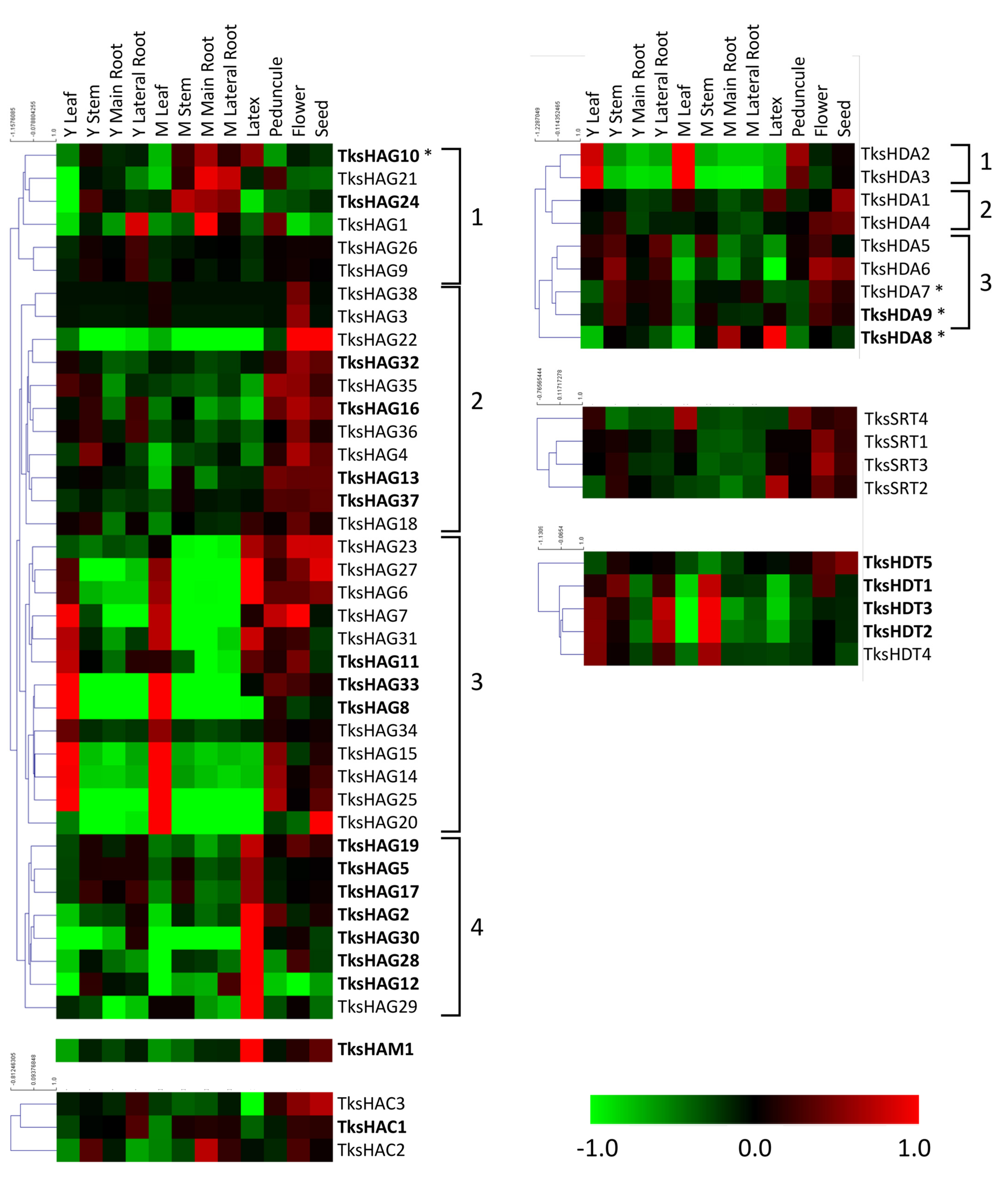
2.2.3. HATs
2.2.4. HDACs
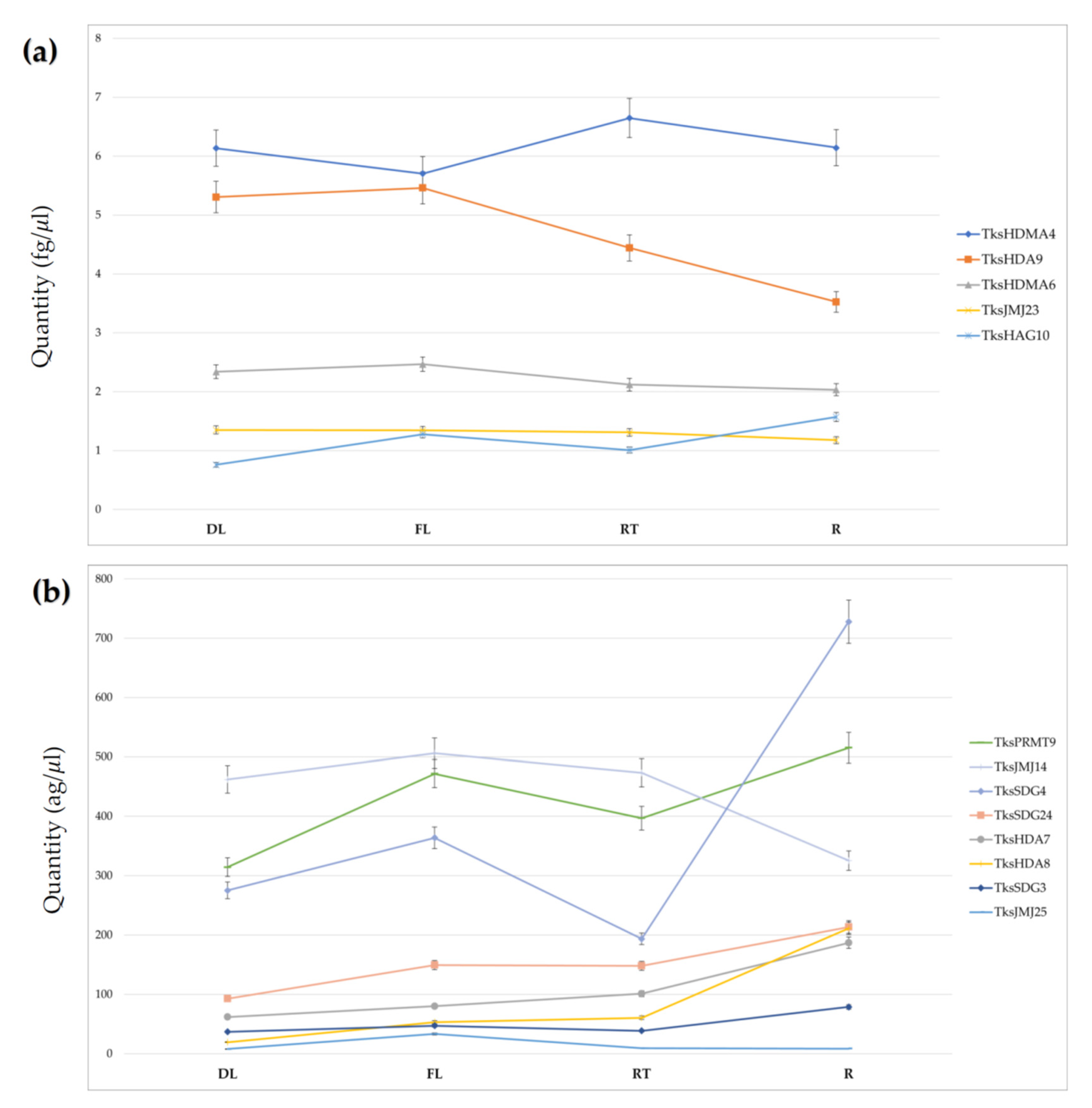
2.3. HMGs Expression
3. Discussion
3.1. HMG Members in Taraxacum Kok-Saghyz
3.2. In Silico and Spatial Analysis of HMGs in Tks Tissues Identified Putative Regulators of NR Biosynthesis
4. Materials and Methods
4.1. Plant Material
4.2. In Silico Identification and Analysis of HMG Loci
4.3. Phylogenetic Analysis
4.4. Transcriptome Analysis
4.5. RNA Extraction, cDNA Synthesis, and Gene Expression Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharifi-Rad, M.; Roberts, T.H.; Matthews, K.R.; Bezerra, C.F.; Morais-Braga, M.F.B.; Coutinho, H.D.M.; Sharopov, F.; Salehi, B.; Yousaf, Z.; Sharifi-Rad, M.; et al. Ethnobotany of the Genus Taraxacum—Phytochemicals and Antimicrobial Activity. Phytother. Res. 2018, 32, 2131–2145. [Google Scholar] [CrossRef]
- Schütz, K.; Carle, R.; Schieber, A. Taraxacum-A Review on Its Phytochemical and Pharmacological Profile. J. Ethnopharmacol. 2006, 107, 313–323. [Google Scholar] [CrossRef]
- Van Beilen, J.B.; Poirier, Y. Establishment of New Crops for the Production of Natural Rubber. Trends Biotechnol. 2007, 25, 522–529. [Google Scholar] [CrossRef]
- Kirschner, J.; Štěpánek, J.; Černý, T.; De Heer, P.; van Dijk, P.J. Available Ex Situ Germplasm of the Potential Rubber Crop Taraxacum Koksaghyz Belongs to a Poor Rubber Producer, T. Brevicorniculatum (Compositae-Crepidinae). Genet. Resour. Crop Evol. 2013, 60, 455–471. [Google Scholar] [CrossRef]
- Mooibroek, H.; Cornish, K. Alternative Sources of Natural Rubber. Appl. Microbiol. Biotechnol. 2000, 53, 355–365. [Google Scholar] [CrossRef]
- Zhang, Y.; Iaffaldano, B.J.; Xie, W.; Blakeslee, J.J.; Cornish, K. Rapid and Hormone-Free Agrobacterium Rhizogenes-Mediated Transformation in Rubber Producing Dandelions Taraxacum Kok-Saghyz and T. Brevicorniculatum. Ind. Crops Prod. 2015, 66, 110–118. [Google Scholar] [CrossRef]
- Panara, F.; Lopez, L.; Daddiego, L.; Fantini, E.; Facella, P.; Perrotta, G. Comparative Transcriptomics between High and Low Rubber Producing Taraxacum Kok-Saghyz R. Plants. BMC Genom. 2018, 19, 875. [Google Scholar] [CrossRef]
- Yamashita, S.; Takahashi, S. Molecular Mechanisms of Natural Rubber Biosynthesis. Annu. Rev. Biochem. 2020, 89, 821–851. [Google Scholar] [CrossRef]
- Lin, T.; Xu, X.; Ruan, J.; Liu, S.; Wu, S.; Shao, X.; Wang, X.; Gan, L.; Qin, B.; Yang, Y.; et al. Genome Analysis of Taraxacum Kok-Saghyz Rodin Provides New Insights into Rubber Biosynthesis. Natl. Sci. Rev. 2018, 5, 78–87. [Google Scholar] [CrossRef]
- Junkong, P.; Ikeda, Y. 7—Properties of Natural Rubbers from Guayule and Rubber Dandelion. In Chemistry, Manufacture, and Application of Natural Rubber, 2nd ed.; Kohjiya, S., Ikeda, Y., Eds.; Woodhead Publishing in Materials; Woodhead Publishing: Sawston, UK, 2021; pp. 177–201. ISBN 978-0-12-818843-9. [Google Scholar]
- Chadwick, M.; Trewin, H.; Gawthrop, F.; Wagstaff, C. Sesquiterpenoids Lactones: Benefits to Plants and People. Int. J. Mol. Sci. 2013, 14, 12780–12805. [Google Scholar] [CrossRef]
- Arias, M.; Herrero, J.; Ricobaraza, M.; Hernández, M.; Ritter, E. Evaluation of Root Biomass, Rubber and Inulincontents in Nine Taraxacum Koksaghyz Rodin Populations. Ind. Crops Prod. 2016, 83, 316–321. [Google Scholar] [CrossRef]
- Esatbeyoglu, T.; Obermair, B.; Dorn, T.; Siems, K.; Rimbach, G.; Birringer, M. Sesquiterpene Lactone Composition and Cellular Nrf2 Induction of Taraxacum Officinale Leaves and Roots and Taraxinic Acid β-d-Glucopyranosyl Ester. J. Med. Food 2017, 20, 71–78. [Google Scholar] [CrossRef]
- Perrella, G.; Kaiserli, E. Light behind the Curtain: Photoregulation of Nuclear Architecture and Chromatin Dynamics in Plants. New Phytol. 2016, 212, 908–919. [Google Scholar] [CrossRef]
- Perrella, G.; Bäurle, I.; van Zanten, M. Epigenetic Regulation of Thermomorphogenesis and Heat Stress Tolerance. New Phytol. 2022, 234, 1144–1160. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Barneche, F.; Malapeira, J.; Mas, P. The Impact of Chromatin Dynamics on Plant Light Responses and Circadian Clock Function. J. Exp. Bot. 2014, 65, 2895–2913. [Google Scholar] [CrossRef]
- Liu, S.; de Jonge, J.; Trejo-Arellano, M.S.; Santos-González, J.; Köhler, C.; Hennig, L. Role of H1 and DNA Methylation in Selective Regulation of Transposable Elements during Heat Stress. New Phytol. 2021, 229, 2238–2250. [Google Scholar] [CrossRef]
- Baumbusch, L.O.; Thorstensen, T.; Krauss, V.; Fischer, A.; Naumann, K.; Assalkhou, R.; Schulz, I.; Reuter, G.; Aalen, R.B. The Arabidopsis Thaliana Genome Contains at Least 29 Active Genes Encoding SET Domain Proteins That Can Be Assigned to Four Evolutionarily Conserved Classes. Nucleic Acids Res. 2001, 29, 4319–4333. [Google Scholar] [CrossRef]
- Zhao, Z.; Shen, W.H. Plants Contain a High Number of Proteins Showing Sequence Similarity to the Animal SUV39H Family of Histone Methyltransferases; New York Academy of Sciences: New York, NY, USA, 2004; Volume 1030, pp. 661–669. [Google Scholar]
- Ng, D.W.-K.; Wang, T.; Chandrasekharan, M.B.; Aramayo, R.; Kertbundit, S.; Hall, T.C. Plant SET Domain-Containing Proteins: Structure, Function and Regulation. Biochim. Biophys. Acta 2007, 1769, 316. [Google Scholar] [CrossRef]
- Ahmad, A.; Dong, Y.; Cao, X. Characterization of the PRMT Gene Family in Rice Reveals Conservation of Arginine Methylation. PLoS ONE 2011, 6, e22664. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Hung, F.-Y.; Yang, S.; Liu, X.; Wu, K. Histone Lysine Demethylases and Their Functions in Plants. Plant Mol. Biol. Rep. 2014, 32, 558–565. [Google Scholar] [CrossRef]
- Schneider, J.; Shilatifard, A. Histone Demethylation by Hydroxylation: Chemistry in Action. ACS Chem. Biol. 2006, 1, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Wang, J.; Lei, C.; Gao, C.; Yang, Y.; Li, Y.; An, N.; Zhang, D.; Han, M. Identification and Characterization of Histone Modification Gene Family Reveal Their Critical Responses to Flower Induction in Apple. BMC Plant Biol. 2018, 18, 173. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N. Removal of H3K27me3 by JMJ Proteins Controls Plant Development and Environmental Responses in Arabidopsis. Front. Plant Sci. 2021, 12, 1212. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Zhang, Y. Regulation of Histone Methylation by Demethylimination and Demethylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 307–318. [Google Scholar] [CrossRef]
- Asensi-Fabado, M.-A.; Amtmann, A.; Perrella, G. Plant Responses to Abiotic Stress: The Chromatin Context of Transcriptional Regulation. Biochim. Biophys. Acta—Gene Regul. Mech. 2017, 1860, 106–122. [Google Scholar] [CrossRef]
- Pandey, R.; Müller, A.; Napoli, C.A.; Selinger, D.A.; Pikaard, C.S.; Richards, E.J.; Bender, J.; Mount, D.W.; Jorgensen, R.A. Analysis of Histone Acetyltransferase and Histone Deacetylase Families of Arabidopsis Thaliana Suggests Functional Diversification of Chromatin Modification among Multicellular Eukaryotes. Nucleic Acids Res. 2002, 30, 5036–5055. [Google Scholar] [CrossRef]
- Servet, C.; Conde e Silva, N.; Zhou, D.-X. Histone Acetyltransferase AtGCN5/HAG1 Is a Versatile Regulator of Developmental and Inducible Gene Expression in Arabidopsis. Mol. Plant 2010, 3, 670–677. [Google Scholar] [CrossRef]
- Tahir, M.S.; Tian, L. HD2-Type Histone Deacetylases: Unique Regulators of Plant Development and Stress Responses. Plant Cell Rep. 2021, 40, 1603–1615. [Google Scholar] [CrossRef]
- Yruela, I.; Moreno-Yruela, C.; Olsen, C.A. Zn2+-Dependent Histone Deacetylases in Plants: Structure and Evolution. Trends Plant Sci. 2021, 26, 741–757. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Liu, X.; Singh, P.; Cui, Y.; Zimmerli, L.; Wu, K. Chromatin Modifications and Remodeling in Plant Abiotic Stress Responses. Biochim. Biophys. Acta—Gene Regul. Mech. 2012, 1819, 129–136. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Tanaka, M.; Kikuchi, A.; Kamada, H. The Arabidopsis Histone Deacetylases HDA6 and HDA19 Contribute to the Repression of Embryonic Properties after Germination. Plant Physiol. 2008, 146, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Krogan, N.T.; Hogan, K.; Long, J.A. APETALA2 Negatively Regulates Multiple Floral Organ Identity Genes in Arabidopsis by Recruiting the Co-Repressor TOPLESS and the Histone Deacetylase HDA19. Dev. Camb. 2012, 139, 4180–4190. [Google Scholar] [CrossRef]
- De Rooij, P.G.H.; Perrella, G.; Kaiserli, E.; van Zanten, M. The Diverse and Unanticipated Roles of Histone Deacetylase 9 in Coordinating Plant Development and Environmental Acclimation. J. Exp. Bot. 2020, 71, 6211–6225. [Google Scholar] [CrossRef] [PubMed]
- Van Der Woude, L.C.; Perrella, G.; Snoek, B.L.; Van Hoogdalem, M.; Novák, O.; Van Verk, M.C.; Van Kooten, H.N.; Zorn, L.E.; Tonckens, R.; Dongus, J.A.; et al. HISTONE DEACETYLASE 9 Stimulates Auxin-Dependent Thermomorphogenesis in Arabidopsis Thaliana by Mediating H2A.Z Depletion. Proc. Natl. Acad. Sci. USA 2019, 116, 25343–25354. [Google Scholar] [CrossRef]
- Shen, R.; Tao, L.; Yang, B. Techno-Economic Analysis of Jet-Fuel Production from Biorefinery Waste Lignin. Biofuels Bioprod. Biorefining 2019, 13, 486–501. [Google Scholar] [CrossRef]
- Jing, Y.; Guo, Q.; Lin, R. The SNL-HDA19 Histone Deacetylase Complex Antagonizes HY5 Activity to Repress Photomorphogenesis in Arabidopsis. New Phytol. 2021, 229, 3221–3236. [Google Scholar] [CrossRef]
- Chen, L.T.; Wu, K. Role of Histone Deacetylases HDA6 and HDA19 in ABA and Abiotic Stress Response. Plant Signal. Behav. 2010, 5, 1318–1320. [Google Scholar] [CrossRef]
- The Arabidopsis Genome Initiative. Analysis of the Genome Sequence of the Flowering Plant Arabidopsis Thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.; Perrella, G.; Calderini, O.; Porceddu, A.; Panara, F. Genome-Wide Identification of Histone Modification Gene Families in the Model Legume Medicago Truncatula and Their Expression Analysis in Nodules. Plants 2022, 11, 322. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Ying, P.; Liu, X.; Li, C.; Xia, R.; Li, J.; Zhao, M. Genome-Wide Identification of Histone Modifiers and Their Expression Patterns during Fruit Abscission in Litchi. Front. Plant Sci. 2017, 8, 639. [Google Scholar] [CrossRef]
- Springer, N.M.; Napoli, C.A.; Selinger, D.A.; Pandey, R.; Cone, K.C.; Chandler, V.L.; Kaeppler, H.F.; Kaeppler, S.M. Comparative Analysis of SET Domain Proteins in Maize and Arabidopsis Reveals Multiple Duplications Preceding the Divergence of Monocots and Dicots. Plant Physiol. 2003, 132, 907–925. [Google Scholar] [CrossRef] [PubMed]
- Aiese Cigliano, R.; Sanseverino, W.; Cremona, G.; Ercolano, M.R.; Conicella, C.; Consiglio, F.M. Genome-Wide Analysis of Histone Modifiers in Tomato: Gaining an Insight into Their Developmental Roles. BMC Genom. 2013, 14, 57. [Google Scholar] [CrossRef]
- Xu, J.; Xu, H.; Liu, Y.; Wang, X.; Xu, Q.; Deng, X. Genome-Wide Identification of Sweet Orange (Citrus Sinensis) Histone Modification Gene Families and Their Expression Analysis during the Fruit Development and Fruit-Blue Mold Infection Process. Front. Plant Sci. 2015, 6, 607. [Google Scholar] [CrossRef]
- Wang, L.; Ahmad, B.; Liang, C.; Shi, X.; Sun, R.; Zhang, S.; Du, G. Bioinformatics and Expression Analysis of Histone Modification Genes in Grapevine Predict Their Involvement in Seed Development, Powdery Mildew Resistance, and Hormonal Signaling. BMC Plant Biol. 2020, 20, 412. [Google Scholar] [CrossRef]
- Zhou, M.; Wan, G.; Mou, P.; Teng, S.; Lin, S.; Wang, G. CNT@NiO/Natural Rubber with Excellent Impedance Matching and Low Interfacial Thermal Resistance toward Flexible and Heat-Conducting Microwave Absorption Applications. J. Mater. Chem. C 2021, 9, 869–880. [Google Scholar] [CrossRef]
- Tran, D.M.; Clément-Demange, A.; Déon, M.; Garcia, D.; Le Guen, V.; Clément-Vidal, A.; Soumahoro, M.; Masson, A.; Label, P.; Le, M.T.; et al. Genetic Determinism of Sensitivity to Corynespora Cassiicola Exudates in Rubber Tree (Hevea Brasiliensis). PLoS ONE 2016, 11, e0162807. [Google Scholar] [CrossRef]
- Parisi, C.A.S.; Kelly, K.J.; Ansotegui, I.J.; Gonzalez-Díaz, S.N.; Bilò, M.B.; Cardona, V.; Park, H.S.; Braschi, M.C.; Macias-Weinmann, A.; Piga, M.A.; et al. Update on Latex Allergy: New Insights into an Old Problem. World Allergy Organ. J. 2021, 14, 100569. [Google Scholar] [CrossRef]
- Hodgson-Kratky, K.J.M.; Stoffyn, O.M.; Wolyn, D.J. Recurrent Selection for Rubber Yield in Russian Dandelion. J. Am. Soc. Hortic. Sci. 2017, 142, 470–475. [Google Scholar] [CrossRef]
- Salehi, M.; Bahmankar, M.; Naghavi, M.R.; Cornish, K. Rubber and Latex Extraction Processes for Taraxacum Kok-Saghyz. Ind. Crops Prod. 2022, 178, 114562. [Google Scholar] [CrossRef]
- Mladenov, V.; Fotopoulos, V.; Kaiserli, E.; Karalija, E.; Maury, S.; Baranek, M.; Segal, N.; Testillano, P.S.; Vassileva, V.; Pinto, G.; et al. Deciphering the Epigenetic Alphabet Involved in Transgenerational Stress Memory in Crops. Int. J. Mol. Sci. 2021, 22, 7118. [Google Scholar] [CrossRef]
- Boyer, L.A.; Latek, R.R.; Peterson, C.L. The SANT Domain: A Unique Histone-Tail-Binding Module? Nat. Rev. Mol. Cell Biol. 2004, 5, 158–163. [Google Scholar] [CrossRef]
- Heo, J.B.; Sung, S. Vernalization-Mediated Epigenetic Silencing by a Long Intronic Noncoding RNA. Science 2011, 331, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, Y.; Liang, Y.; Zhou, D.; Li, S.; Lin, S.; Dong, H.; Huang, L. The Function of Histone Lysine Methylation Related SET Domain Group Proteins in Plants. Protein Sci. 2020, 29, 1120–1137. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-G.; Ye, Y. Bag6/Bat3/Scythe: A Novel Chaperone Activity with Diverse Regulatory Functions in Protein Biogenesis and Degradation. BioEssays 2013, 35, 377–385. [Google Scholar] [CrossRef]
- Dong, Y.; Lu, J.; Liu, J.; Jalal, A.; Wang, C. Genome-Wide Identification and Functional Analysis of JmjC Domain-Containing Genes in Flower Development of Rosa Chinensis. Plant Mol. Biol. 2020, 102, 417–430. [Google Scholar] [CrossRef]
- Hoo, S.C.; Howe, G.A. A Critical Role for the TIFY Motif in Repression of Jasmonate Signaling by a Stabilized Splice Variant of the JASMONATE ZIM-Domain Protein JAZ10 in Arabidopsis. Plant Cell 2009, 21, 131–145. [Google Scholar] [CrossRef]
- Kanyal, A.; Rawat, M.; Gurung, P.; Choubey, D.; Anamika, K.; Karmodiya, K. Genome-Wide Survey and Phylogenetic Analysis of Histone Acetyltransferases and Histone Deacetylases of Plasmodium Falciparum. FEBS J. 2018, 285, 1767–1782. [Google Scholar] [CrossRef]
- Chen, C.Y.; Wu, K.; Schmidt, W. The Histone Deacetylase HDA19 Controls Root Cell Elongation and Modulates a Subset of Phosphate Starvation Responses in Arabidopsis. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Lenders, M.; Hillebrand, A.; van Deenen, N.; Munt, O.; Reichelt, R.; Eisenreich, W.; Fischer, R.; Prüfer, D.; Schulze Gronover, C. Characterization of Rubber Particles and Rubber Chain Elongation in Taraxacum Koksaghyz. BMC Biochem. 2010, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- De Lucia, F.; Crevillen, P.; Jones, A.M.E.; Greb, T.; Dean, C. A PHD-Polycomb Repressive Complex 2 Triggers the Epigenetic Silencing of FLC during Vernalization. Proc. Natl. Acad. Sci. USA 2008, 105, 16831–16836. [Google Scholar] [CrossRef]
- Berr, A.; McCallum, E.J.; Ménard, R.; Meyer, D.; Fuchs, J.; Dong, A.; Shen, W.H. Arabidopsis SET DOMAIN GROUP2 Is Required for H3K4 Trimethylation and Is Crucial for Both Sporophyte and Gametophyte Development. Plant Cell 2010, 22, 3232–3248. [Google Scholar] [CrossRef] [PubMed]
- Crevillén, P.; Yang, H.; Cui, X.; Greeff, C.; Trick, M.; Qiu, Q.; Cao, X.; Dean, C. Epigenetic Reprogramming That Prevents Transgenerational Inheritance of the Vernalized State. Nature 2014, 515, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. 20 Years of the SMART Protein Domain Annotation Resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent Updates, New Developments and Status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An Upgraded Gene Feature Visualization Server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A Toolkit for Detection and Evolutionary Analysis of Gene Synteny and Collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Löytynoja, A.; Goldman, N. An Algorithm for Progressive Multiple Alignment of Sequences with Insertions. Proc. Natl. Acad. Sci. USA 2005, 102, 10557–10562. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML: A Program Package for Phylogenetic Analysis by Maximum Likelihood. Bioinformatics 1997, 13, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Estimating Synonymous and Nonsynonymous Substitution Rates Under Realistic Evolutionary Models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Corchete, L.A.; Rojas, E.A.; Alonso-López, D.; De Las Rivas, J.; Gutiérrez, N.C.; Burguillo, F.J. Systematic Comparison and Assessment of RNA-Seq Procedures for Gene Expression Quantitative Analysis. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15. [Google Scholar] [CrossRef]
- Putri, G.H.; Anders, S.; Pyl, P.T.; Pimanda, J.E.; Zanini, F. Analysing High-Throughput Sequencing Data in Python with HTSeq 2.0. Bioinformatics 2022, 38, 2943–2945. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinforma. Oxf. Engl. 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Whelan, J.A.; Russell, N.B.; Whelan, M.A. A Method for the Absolute Quantification of CDNA Using Real-Time PCR. J. Immunol. Methods 2003, 278, 261–269. [Google Scholar] [CrossRef]
- D’Amelia, V.; Aversano, R.; Batelli, G.; Caruso, I.; Moreno, M.C.; Castro-Sanz, A.B.; Chiaiese, P.; Fasano, C.; Palomba, F.; Carputo, D. High AN1 Variability and Interaction with Basic Helix-Loop-Helix Co-Factors Related to Anthocyanin Biosynthesis in Potato Leaves. Plant J. 2014, 80, 527–540. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Group | PFAM | Taraxacum Kok-Saghyz | Lactuca Sativa | Arabidopsis Thaliana | Medicago Truncatula v.4.01 |
|---|---|---|---|---|---|---|
| HMT | SDG | PF00856 | 46 | 46 | 41 | 78 |
| PRMT | PF05185 | 14 | 4 | 7 | 3 | |
| HDM | JMJ | PF02373 | 27 | 47 | 21 | 34 |
| HDMA | PF04433 | 7 | 8 | 4 | 12 | |
| HAT | HAG | PF00583 | 38 | 34 | 3 | 51 |
| HAM | PF01853 | 1 | 2 | 2 | 1 | |
| HAC | PF08214 | 3 | 14 | 5 | 11 | |
| HAF | PF09247 | 0 | 1 | 2 | 1 | |
| HDAC | HDA | PF00850 | 9 | 19 | 12 | 10 |
| SRT | PF02146 | 4 | 3 | 2 | 2 | |
| HDT | 5 | 3 | 4 | 3 | ||
| 154 | 181 | 103 | 206 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panara, F.; Fasano, C.; Lopez, L.; Porceddu, A.; Facella, P.; Fantini, E.; Daddiego, L.; Perrella, G. Genome-Wide Identification and Spatial Expression Analysis of Histone Modification Gene Families in the Rubber Dandelion Taraxacum kok-saghyz. Plants 2022, 11, 2077. https://doi.org/10.3390/plants11162077
Panara F, Fasano C, Lopez L, Porceddu A, Facella P, Fantini E, Daddiego L, Perrella G. Genome-Wide Identification and Spatial Expression Analysis of Histone Modification Gene Families in the Rubber Dandelion Taraxacum kok-saghyz. Plants. 2022; 11(16):2077. https://doi.org/10.3390/plants11162077
Chicago/Turabian StylePanara, Francesco, Carlo Fasano, Loredana Lopez, Andrea Porceddu, Paolo Facella, Elio Fantini, Loretta Daddiego, and Giorgio Perrella. 2022. "Genome-Wide Identification and Spatial Expression Analysis of Histone Modification Gene Families in the Rubber Dandelion Taraxacum kok-saghyz" Plants 11, no. 16: 2077. https://doi.org/10.3390/plants11162077
APA StylePanara, F., Fasano, C., Lopez, L., Porceddu, A., Facella, P., Fantini, E., Daddiego, L., & Perrella, G. (2022). Genome-Wide Identification and Spatial Expression Analysis of Histone Modification Gene Families in the Rubber Dandelion Taraxacum kok-saghyz. Plants, 11(16), 2077. https://doi.org/10.3390/plants11162077