
Spatiotemporal Transcriptomic Atlas of Developing Embryos and Vegetative Tissues in Flax

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Plant Growth Conditions and Tissue Collection
2.2. RNA Extraction and High-Throughput Sequencing
2.3. Mapping of RNA-seq Read to Reference Genome and Analysis of Expressed Genes
2.4. Principal Component Analysis (PCA), Hierarchical Clustering, and Sample Distance
2.5. Gene Co-Expression Analysis and WGCNA Network Construction
2.6. Identification of Tissue-Specific Expressed Genes and Functional Classification
2.7. Gene Correlation and Transcription Factor Regulation Analysis in Different Gene Categories
3. Results
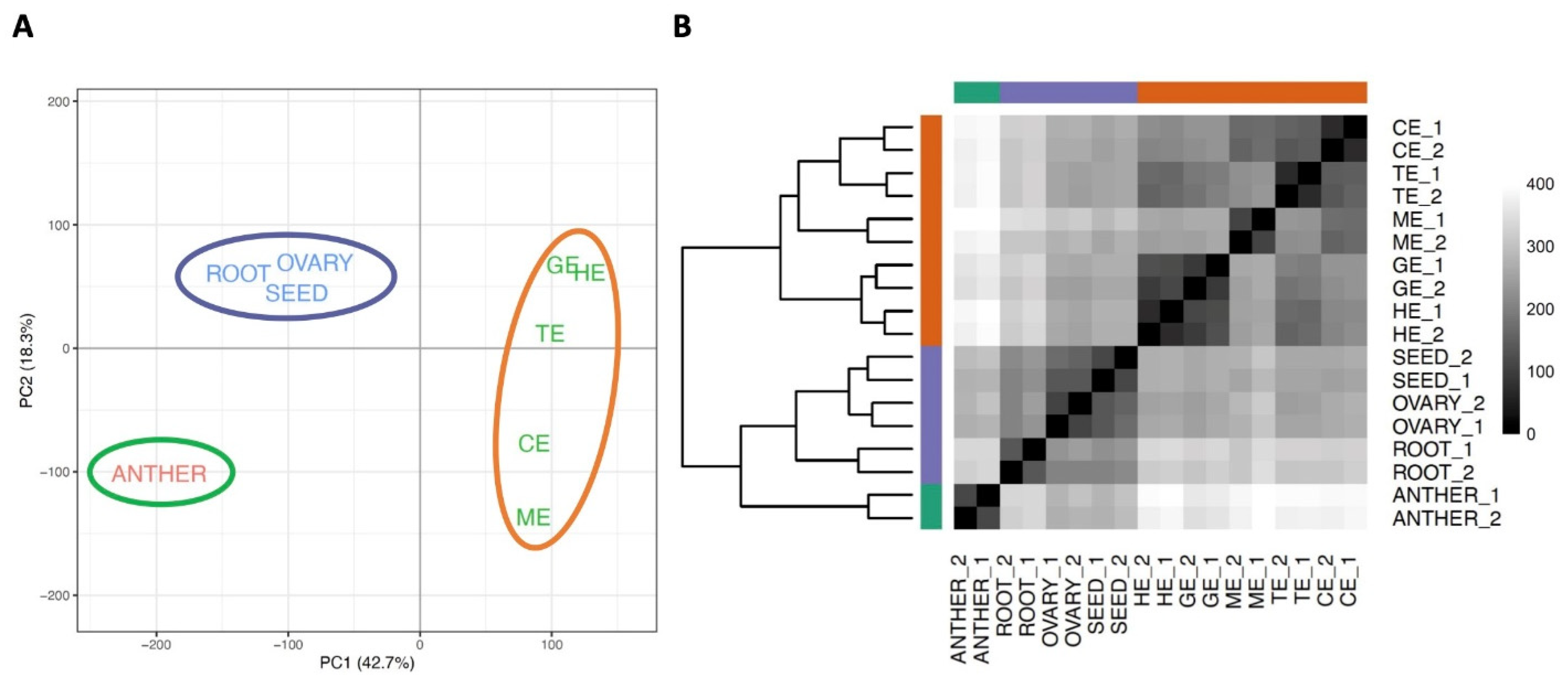
3.1. Transcriptome Profiling of Flax
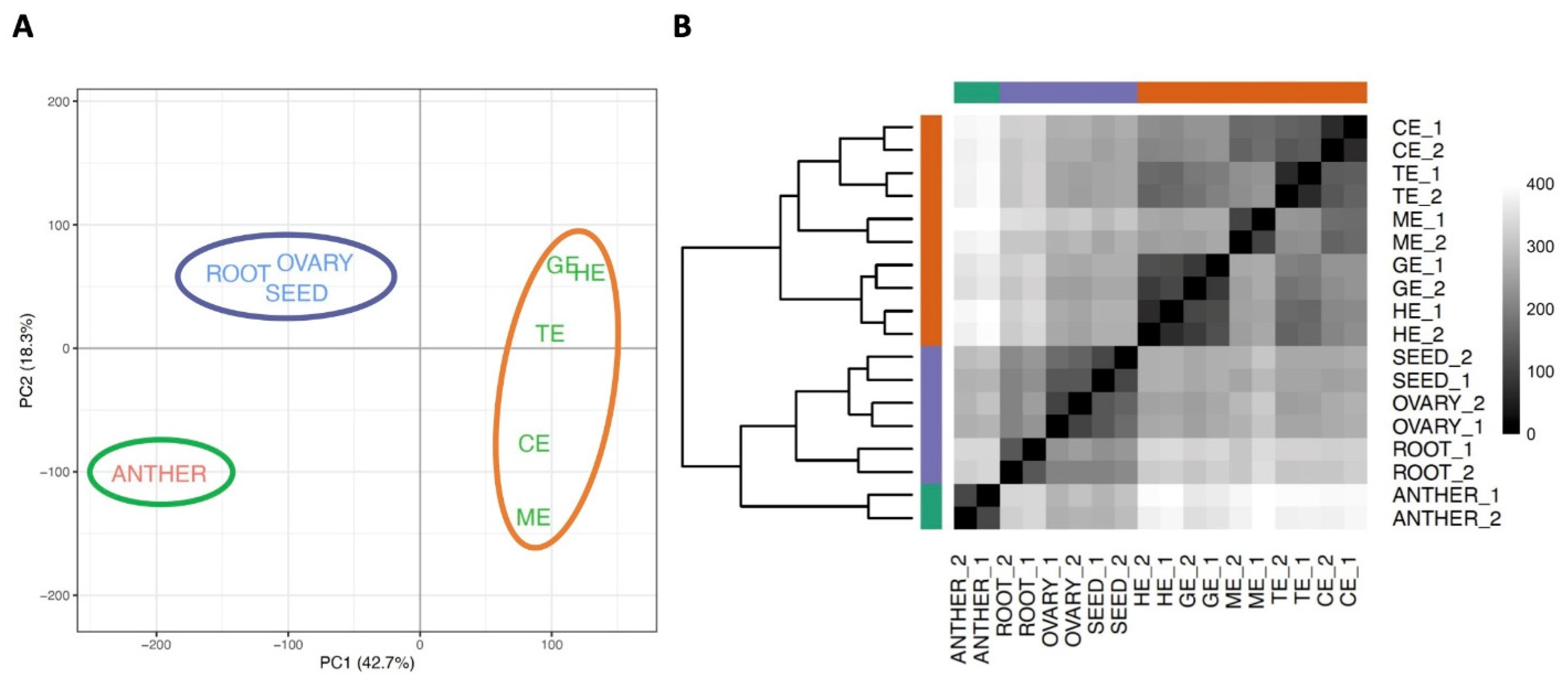
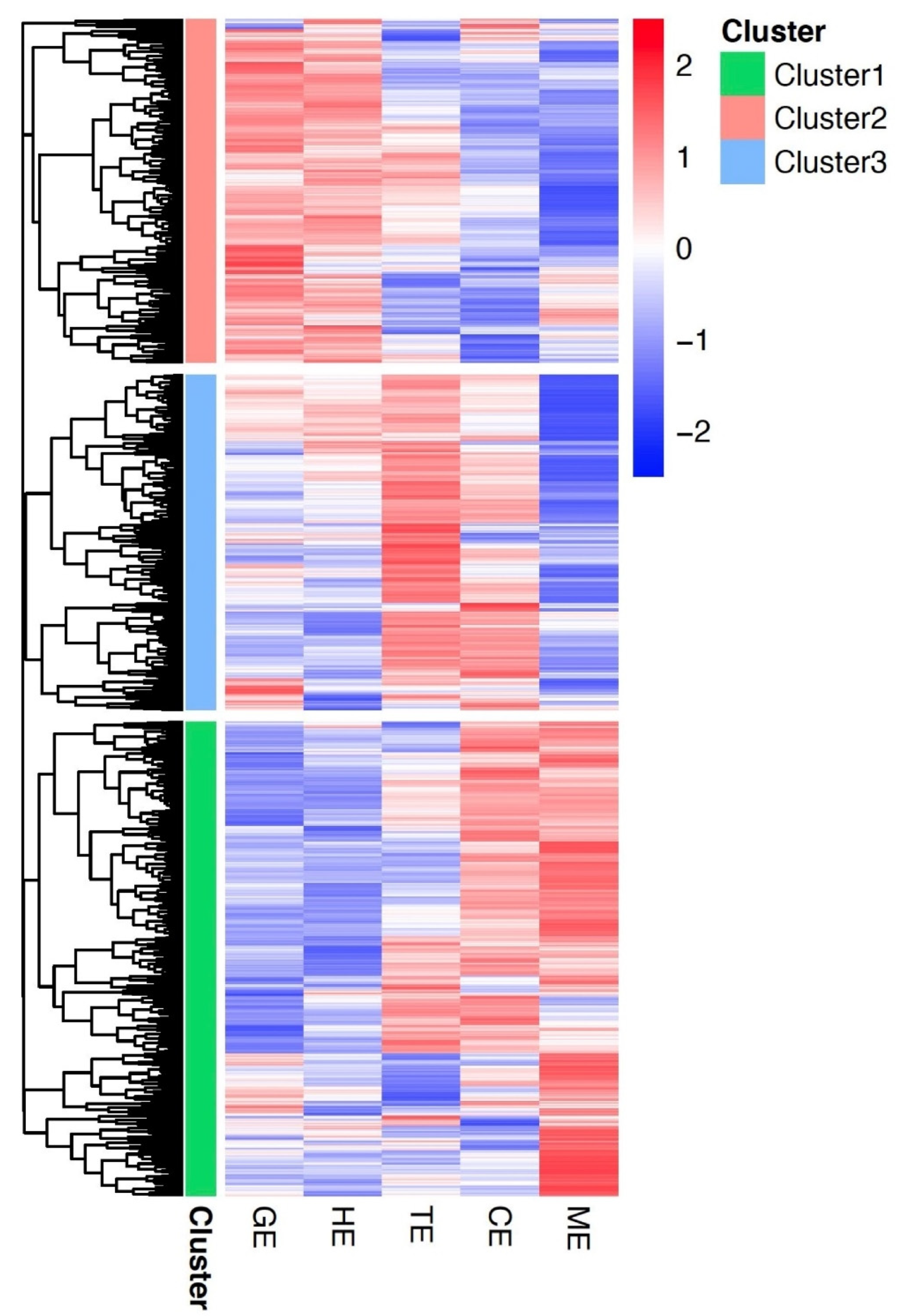
3.2. Relationship of the Transcriptomes in Different Developmental Stages of Flax
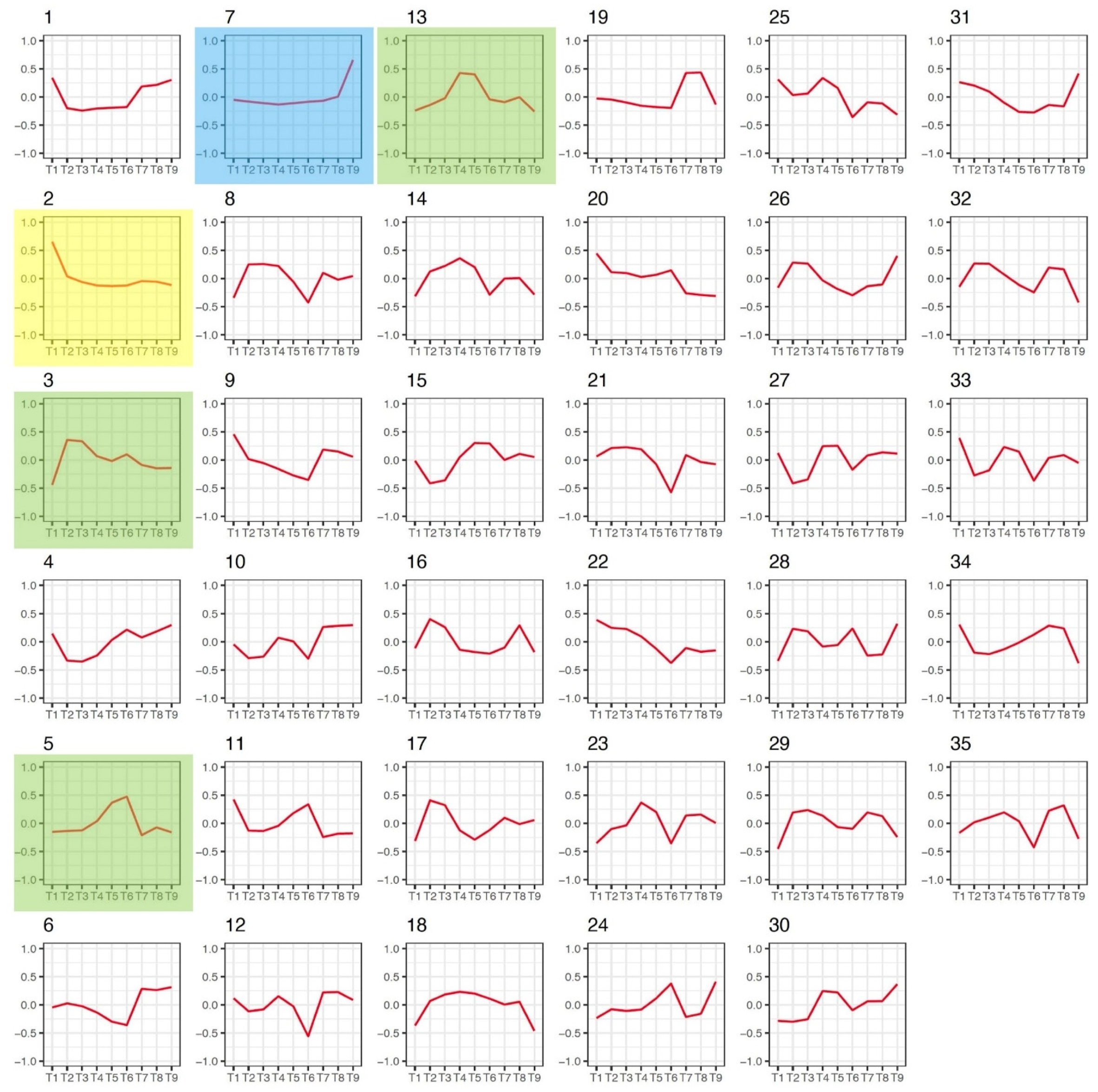
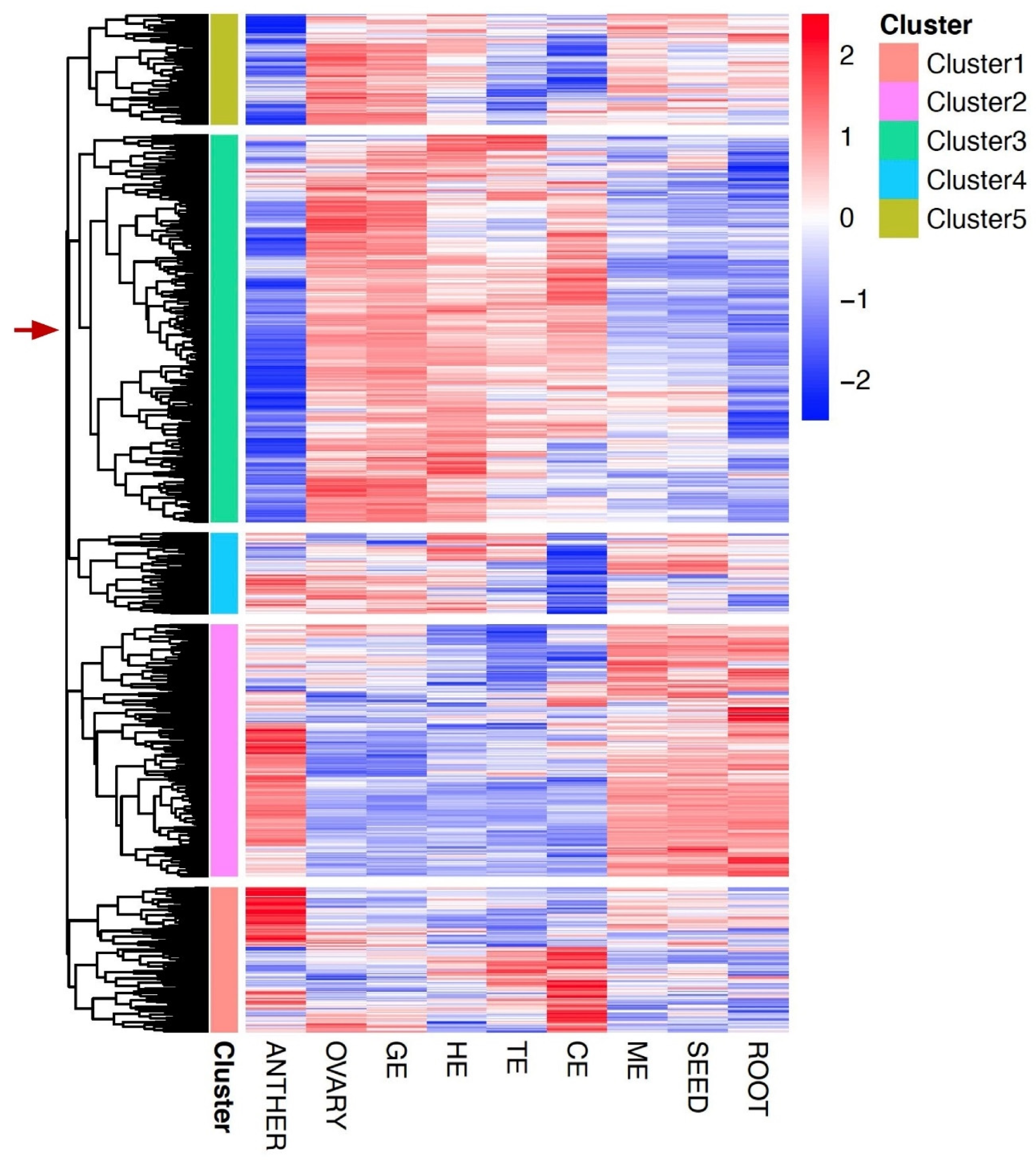
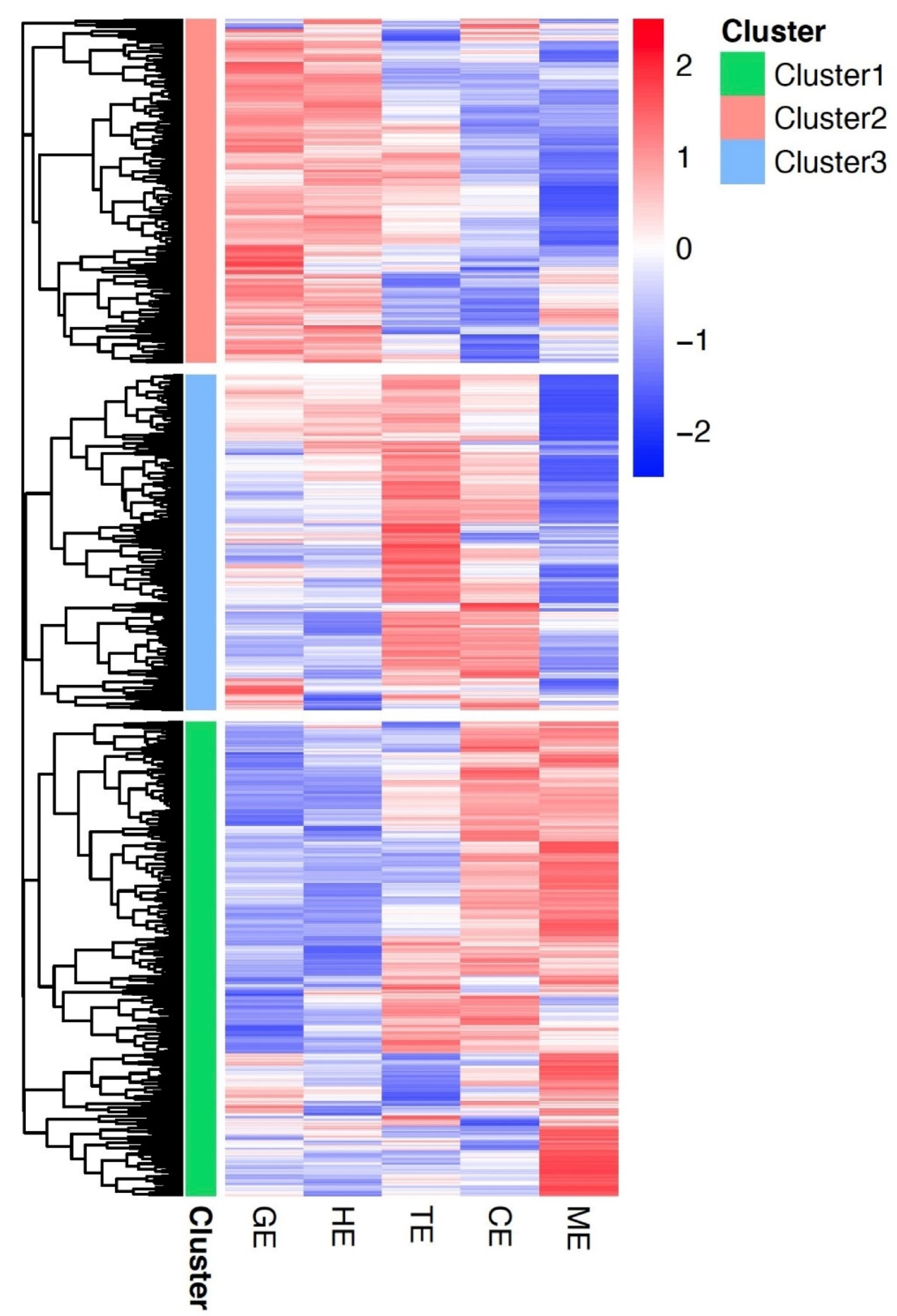
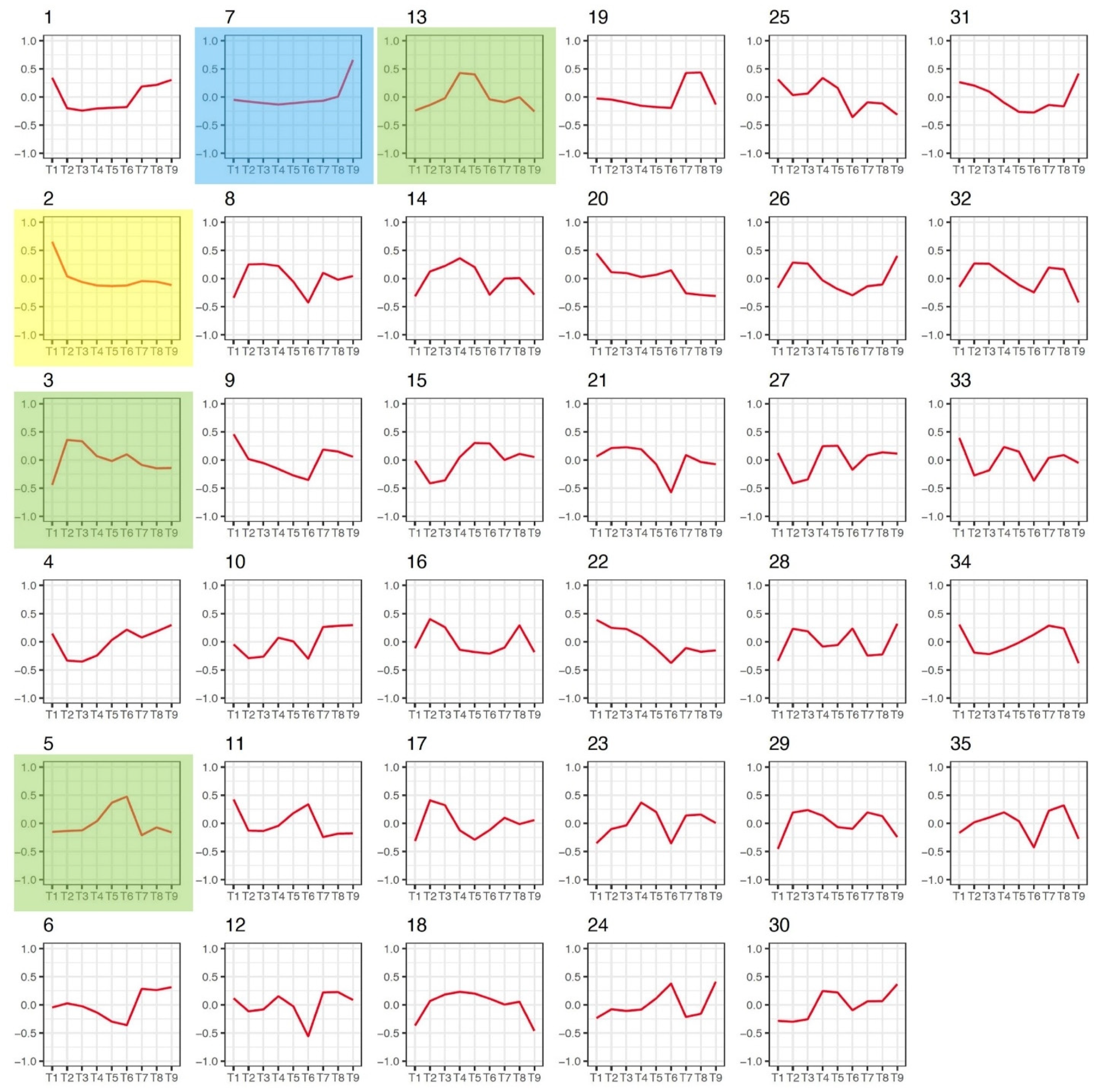
3.3. Gene Co-Expression Networks in Flax
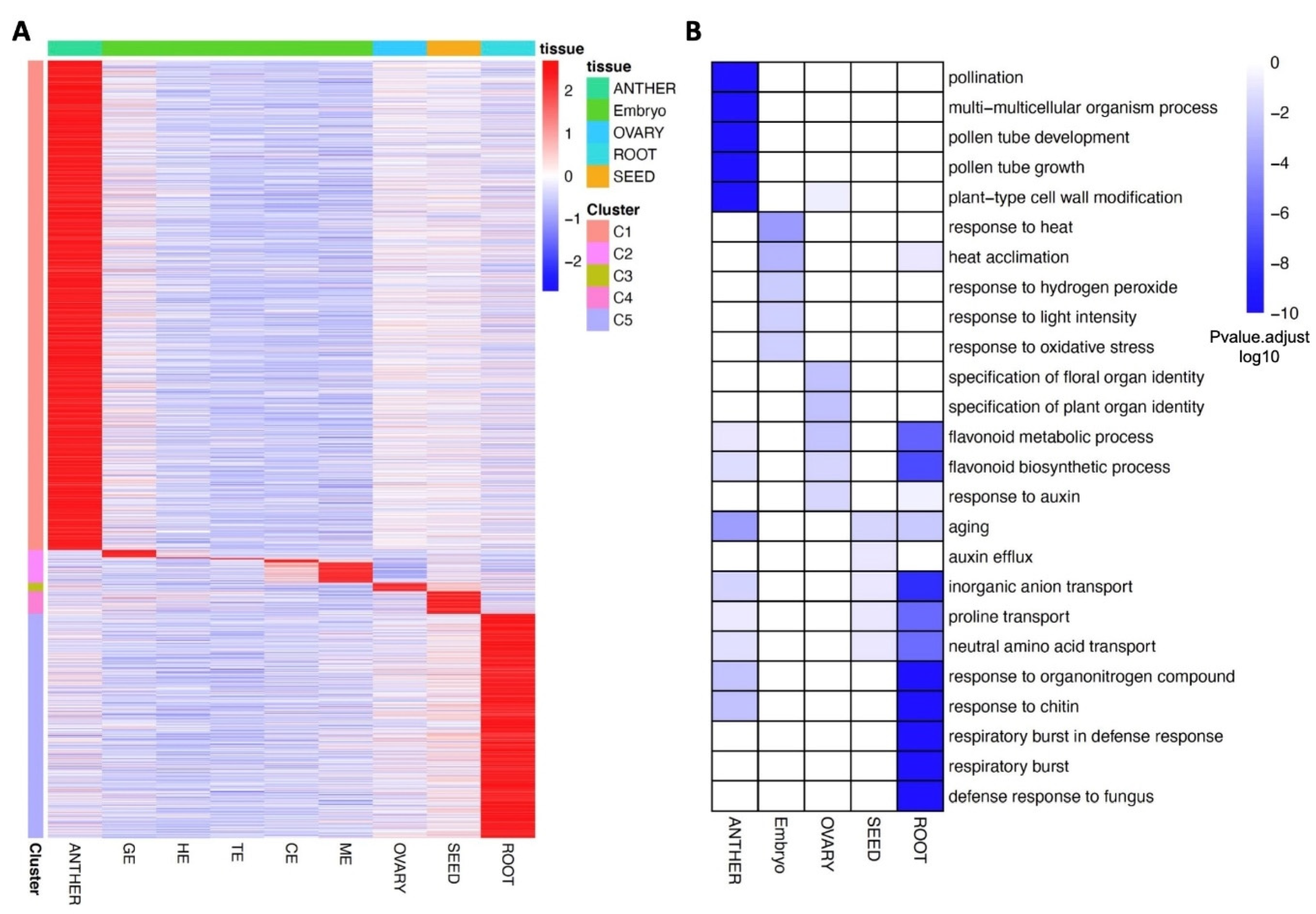
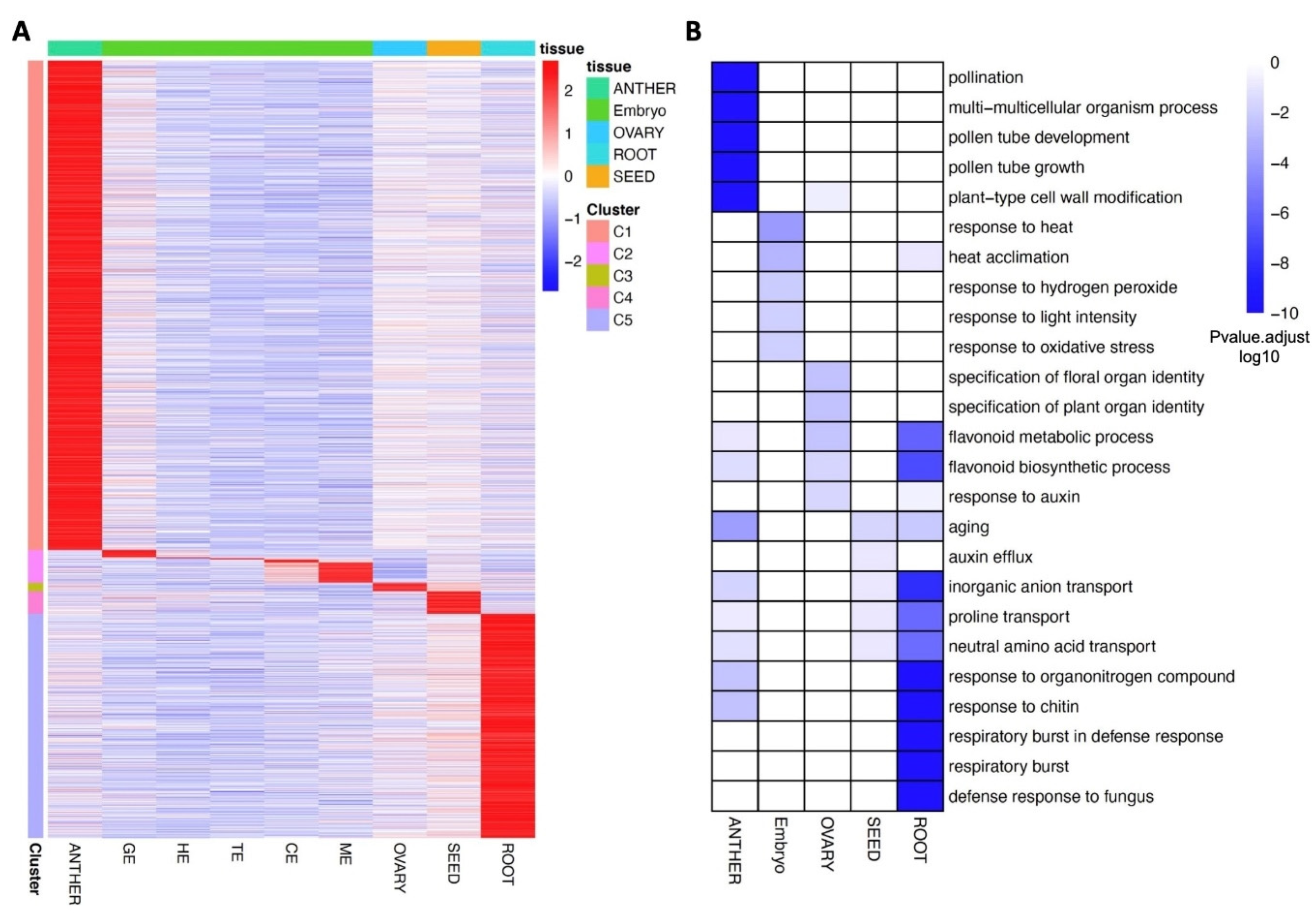
3.4. Dominantly Expressed Genes and Biological Processes in Different Tissues of Flax
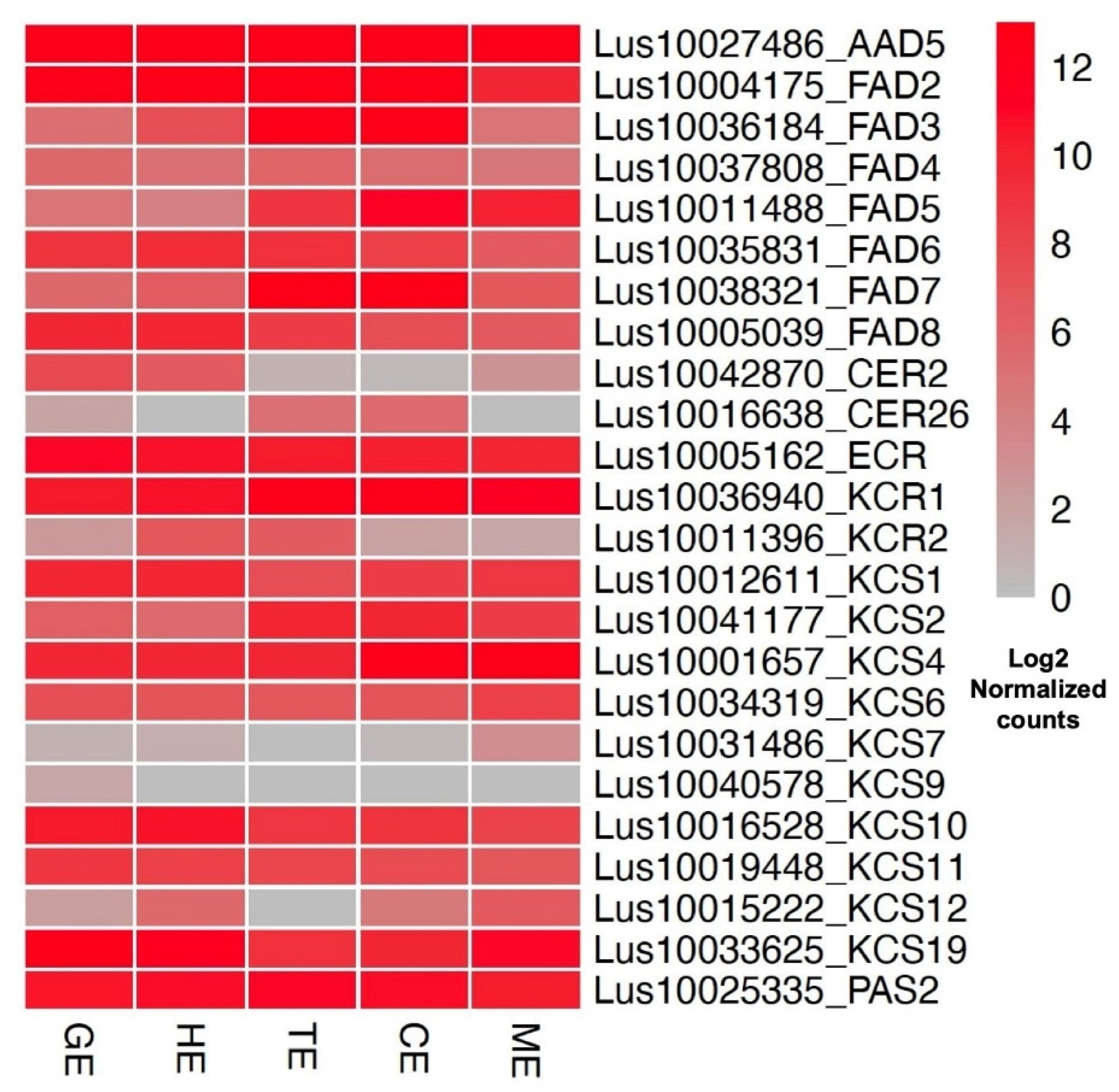
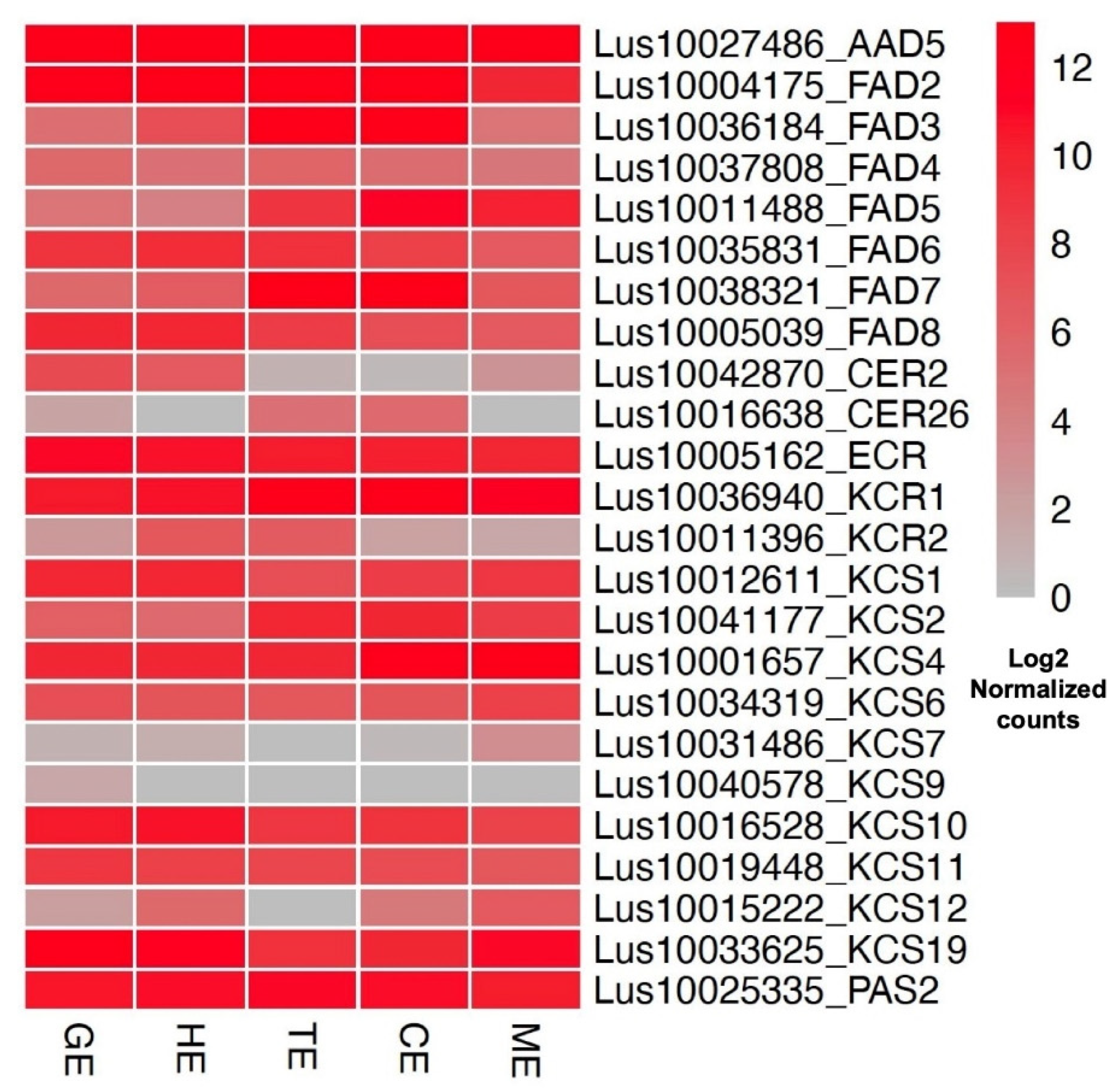
3.5. Expression Profiles of Transcription Factors and Embryo-Development-Essential Genes in Flax
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oomah, B.D. Flaxseed as a functional food source. J. Sci. Food Agric. 2001, 81, 889–894. [Google Scholar] [CrossRef]
- Touré, A.; Xueming, X. Flaxseed Lignans: Source, Biosynthesis, Metabolism, Antioxidant Activity, Bio-Active Components, and Health Benefits. Compr. Rev. Food Sci. Food Saf. 2010, 9, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Mridula, D.; Rehal, J.; Barnwal, P. Flaxseed: A potential source of food, feed and fiber. Crit. Rev. Food Sci. Nutr. 2011, 51, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Sharma, V.; Upadhyay, N.; Gill, S.; Sihag, M. Flax and flaxseed oil: An ancient medicine & modern functional food. J. Food Sci. Technol. 2014, 51, 1633–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, S.; Rashevskaya, T.; Makhonina, M. Flaxseed additive application in dairy products production. Procedia Food Sci. 2011, 1, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Bastaki, N.K.; Cullis, C.A. Floral-dip transformation of flax (Linum usitatissimum) to generate transgenic progenies with a high transformation rate. J. Vis. Exp. JoVE 2014, 94, 52189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Hobson, N.; Galindo, L.; Zhu, S.; Shi, D.; McDill, J.; Yang, L.; Hawkins, S.; Neutelings, G.; Datla, R.; et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. Cell Mol. Biol. 2012, 72, 461–473. [Google Scholar] [CrossRef] [Green Version]
- Dmitriev, A.A.; Kudryavtseva, A.V.; Krasnov, G.S.; Koroban, N.V.; Speranskaya, A.S.; Krinitsina, A.A.; Belenikin, M.S.; Snezhkina, A.V.; Sadritdinova, A.F.; Kishlyan, N.V.; et al. Gene expression profiling of flax (Linum usitatissimum L.) under edaphic stress. BMC Plant Biol. 2016, 16, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, P.K.; Rai, R.; Mahato, A.K.; Gaikwad, K.; Singh, N.K. Transcriptome Landscape at Different Developmental Stages of a Drought Tolerant Cultivar of Flax (Linum usitatissimum). Front. Chem. 2017, 5, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, P.K.; Cao, Y.; Jailani, A.K.; Gupta, P.; Venglat, P.; Xiang, D.; Rai, R.; Sharma, R.; Thirunavukkarasu, N.; Abdin, M.Z.; et al. Genome-wide analysis of drought induced gene expression changes in flax (Linum usitatissimum). GM Crops Food 2014, 5, 106–119. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Deyholos, M.K. RNASeq Analysis of the Shoot Apex of Flax (Linum usitatissimum) to Identify Phloem Fiber Specification Genes. Front. Plant Sci. 2016, 7, 950. [Google Scholar] [CrossRef] [Green Version]
- Gui, B.; Shim, Y.Y.; Datla, R.S.S.; Covello, P.S.; Stone, S.L.; Reaney, M.J.T. Identification and Quantification of Cyclolinopeptides in Five Flaxseed Cultivars. J. Agric. Food Chem. 2012, 60, 8571–8579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, D.; Dai, Z.; Yang, Z.; Tang, Q.; Deng, C.; Xu, Y.; Wang, J.; Chen, J.; Zhao, D.; Zhang, S.; et al. Combined genome-wide association analysis and transcriptome sequencing to identify candidate genes for flax seed fatty acid metabolism. Plant Sci. Int. J. Exp. Plant Biol. 2019, 286, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, Z.; Song, X.; Han, L.; Wang, L.; Zhang, J.; Long, Y.; Pei, X. Identification and Characterization of microRNAs in the Developing Seed of Linseed Flax (Linum usitatissimum L.). Int. J. Mol. Sci. 2020, 21, 2708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barvkar, V.T.; Pardeshi, V.C.; Kale, S.M.; Qiu, S.; Rollins, M.; Datla, R.; Gupta, V.S.; Kadoo, N.Y. Genome-wide identification and characterization of microRNA genes and their targets in flax (Linum usitatissimum): Characterization of flax miRNA genes. Planta 2013, 237, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Venglat, P.; Tibiche, C.; Yang, H.; Risseeuw, E.; Cao, Y.; Babic, V.; Cloutier, M.; Keller, W.; Wang, E.; et al. Genome-Wide Analysis Reveals Gene Expression and Metabolic Network Dynamics during Embryo Development in Arabidopsis. Plant Physiol. 2011, 156, 346–356. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Xiang, D.; Quilichini, T.D.; Venglat, P.; Pandey, P.K.; Wang, E.; Gillmor, C.S.; Datla, R. Gene expression atlas of embryo development in Arabidopsis. Plant Reprod. 2019, 32, 93–104. [Google Scholar] [CrossRef]
- Xiang, D.; Quilichini, T.D.; Liu, Z.; Gao, P.; Pan, Y.; Li, Q.; Nilsen, K.T.; Venglat, P.; Esteban, E.; Pasha, A.; et al. The Transcriptional Landscape of Polyploid Wheats and Their Diploid Ancestors during Embryogenesis and Grain Development. Plant Cell 2019, 31, 2888–2911. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Quilichini, T.D.; Yang, H.; Li, Q.; Nilsen, K.T.; Qin, L.; Babic, V.; Liu, L.; Cram, D.; Pasha, A.; et al. Evolutionary divergence in embryo and seed coat development of U’s Triangle Brassica species illustrated by a spatiotemporal transcriptome atlas. New Phytol. 2022, 233, 30–51. [Google Scholar] [CrossRef]
- Venglat, P.; Xiang, D.; Qiu, S.; Stone, S.L.; Tibiche, C.; Cram, D.; Alting-Mees, M.; Nowak, J.; Cloutier, S.; Deyholos, M.; et al. Gene expression analysis of flax seed development. BMC Plant Biol. 2011, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Kryuchkova-Mostacci, N.; Robinson-Rechavi, M. A benchmark of gene expression tissue-specificity metrics. Brief. Bioinform. 2017, 18, 205–214. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Schwacke, R.; Ponce-Soto, G.Y.; Krause, K.; Bolger, A.M.; Arsova, B.; Hallab, A.; Gruden, K.; Stitt, M.; Bolger, M.E.; Usadel, B. MapMan4: A Refined Protein Classification and Annotation Framework Applicable to Multi-Omics Data Analysis. Mol. Plant 2019, 12, 879–892. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Hao, Z.; Zhang, Z.; Xiang, D.; Venglat, P.; Chen, J.; Gao, P.; Datla, R.; Weijers, D. Conserved, divergent and heterochronic gene expression during Brachypodium and Arabidopsis embryo development. Plant Reprod. 2021, 34, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, M.; Roig-Villanova, I.; Zanchetti, E.; Caselli, F.; Gregis, V.; Bardetti, P.; Chiara, M.; Guazzotti, A.; Caporali, E.; Mendes, M.A.; et al. MADS-Box and bHLH Transcription Factors Coordinate Transmitting Tract Development in Arabidopsis thaliana. Front. Plant Sci. 2020, 11, 526. [Google Scholar] [CrossRef]
- Meinke, D.W. Genome-wide identification of EMBRYO-DEFECTIVE (EMB) genes required for growth and development in Arabidopsis. New Phytol. 2020, 226, 306–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, N.; Wang, Y.; Meinke, D. Natural Variation in Sensitivity to a Loss of Chloroplast Translation in Arabidopsis. Plant Physiol. 2014, 166, 2013–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, M.; Rogers, R.; Muralla, R.; Meinke, D. Requirement of aminoacyl-tRNA synthetases for gametogenesis and embryo development in Arabidopsis. Plant J. Cell Mol. Biol. 2005, 44, 866–878. [Google Scholar] [CrossRef] [PubMed]
- Chepyshko, H.; Lai, C.-P.; Huang, L.-M.; Liu, J.-H.; Shaw, J.-F. Multifunctionality and diversity of GDSL esterase/lipase gene family in rice (Oryza sativa L. japonica) genome: New insights from bioinformatics analysis. BMC Genom. 2012, 13, 309. [Google Scholar] [CrossRef] [Green Version]
- Stone, S.L.; Kwong, L.W.; Yee, K.M.; Pelletier, J.; Lepiniec, L.; Fischer, R.L.; Goldberg, R.B.; Harada, J.J. LEAFY COTYLEDON2 encodes a B3 domain transcription factor that induces embryo development. Proc. Natl. Acad. Sci. USA 2001, 98, 11806–11811. [Google Scholar] [CrossRef] [Green Version]
- Singer, S.D.; Jayawardhane, K.N.; Jiao, C.; Weselake, R.J.; Chen, G. The effect of AINTEGUMENTA-LIKE 7 over-expression on seed fatty acid biosynthesis, storage oil accumulation and the transcriptome in Arabidopsis thaliana. Plant Cell Rep. 2021, 40, 1647–1663. [Google Scholar] [CrossRef]
- Ohlrogge, J.; Browse, J. Lipid biosynthesis. Plant Cell 1995, 7, 957–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Quilichini, T.D.; Zhai, C.; Qin, L.; Nilsen, K.T.; Li, Q.; Sharpe, A.G.; Kochian, L.V.; Zou, J.; Reddy, A.S.N.; et al. Alternative splicing dynamics and evolutionary divergence during embryogenesis in wheat species. Plant Biotechnol. J. 2021, 19, 1624–1643. [Google Scholar] [CrossRef]
- James, D.W., Jr.; Lim, E.; Keller, J.; Plooy, I.; Ralston, E.; Dooner, H.K. Directed tagging of the Arabidopsis FATTY ACID ELONGATION1 (FAE1) gene with the maize transposon activator. Plant Cell 1995, 7, 309–319. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, P.; Qiu, S.; Ma, X.; Parkin, I.A.P.; Xiang, D.; Datla, R. Spatiotemporal Transcriptomic Atlas of Developing Embryos and Vegetative Tissues in Flax. Plants 2022, 11, 2031. https://doi.org/10.3390/plants11152031
Gao P, Qiu S, Ma X, Parkin IAP, Xiang D, Datla R. Spatiotemporal Transcriptomic Atlas of Developing Embryos and Vegetative Tissues in Flax. Plants. 2022; 11(15):2031. https://doi.org/10.3390/plants11152031
Chicago/Turabian StyleGao, Peng, Shuqing Qiu, Xingliang Ma, Isobel A. P. Parkin, Daoquan Xiang, and Raju Datla. 2022. "Spatiotemporal Transcriptomic Atlas of Developing Embryos and Vegetative Tissues in Flax" Plants 11, no. 15: 2031. https://doi.org/10.3390/plants11152031
APA StyleGao, P., Qiu, S., Ma, X., Parkin, I. A. P., Xiang, D., & Datla, R. (2022). Spatiotemporal Transcriptomic Atlas of Developing Embryos and Vegetative Tissues in Flax. Plants, 11(15), 2031. https://doi.org/10.3390/plants11152031