
Sugarcane Genotypes with Contrasting Biological Nitrogen Fixation Efficiencies Differentially Modulate Nitrogen Metabolism, Auxin Signaling, and Microorganism Perception Pathways

 ,
,  ,
,
Abstract
1. Introduction
2. Results and Discussion
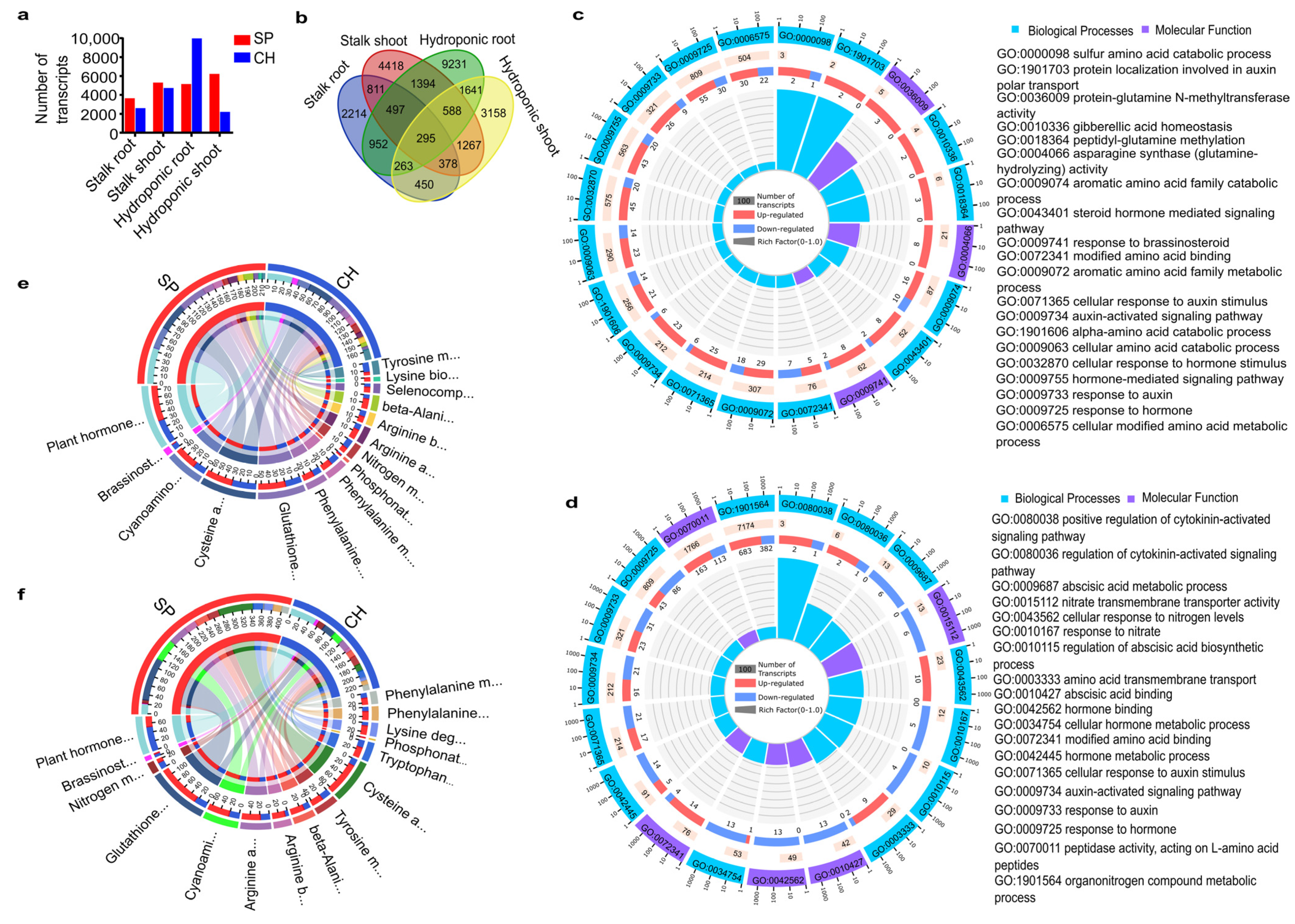
2.1. RNA Sequencing of Sugarcane Genotypes with High and Low BNF Efficiencies
2.2. Differential Expression Profiles in the Two Sugarcane BNF-Contrasting Genotypes
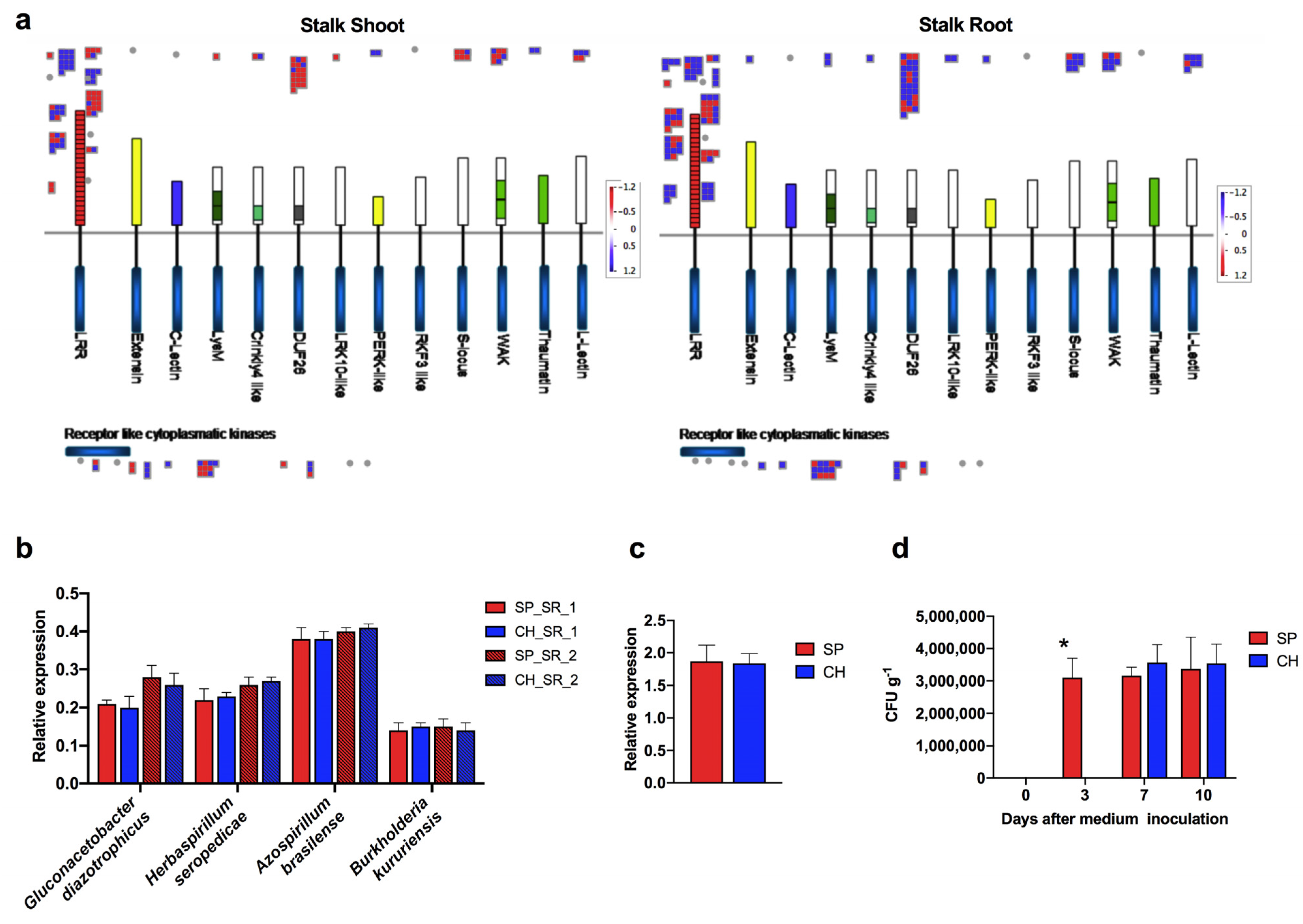
2.3. Plant Receptors Involved in Plant–Bacteria Recognition
2.3.1. NBS-LRR Proteins
2.3.2. LRR-Kinase Family
2.3.3. Endophytic Bacterial Colonization
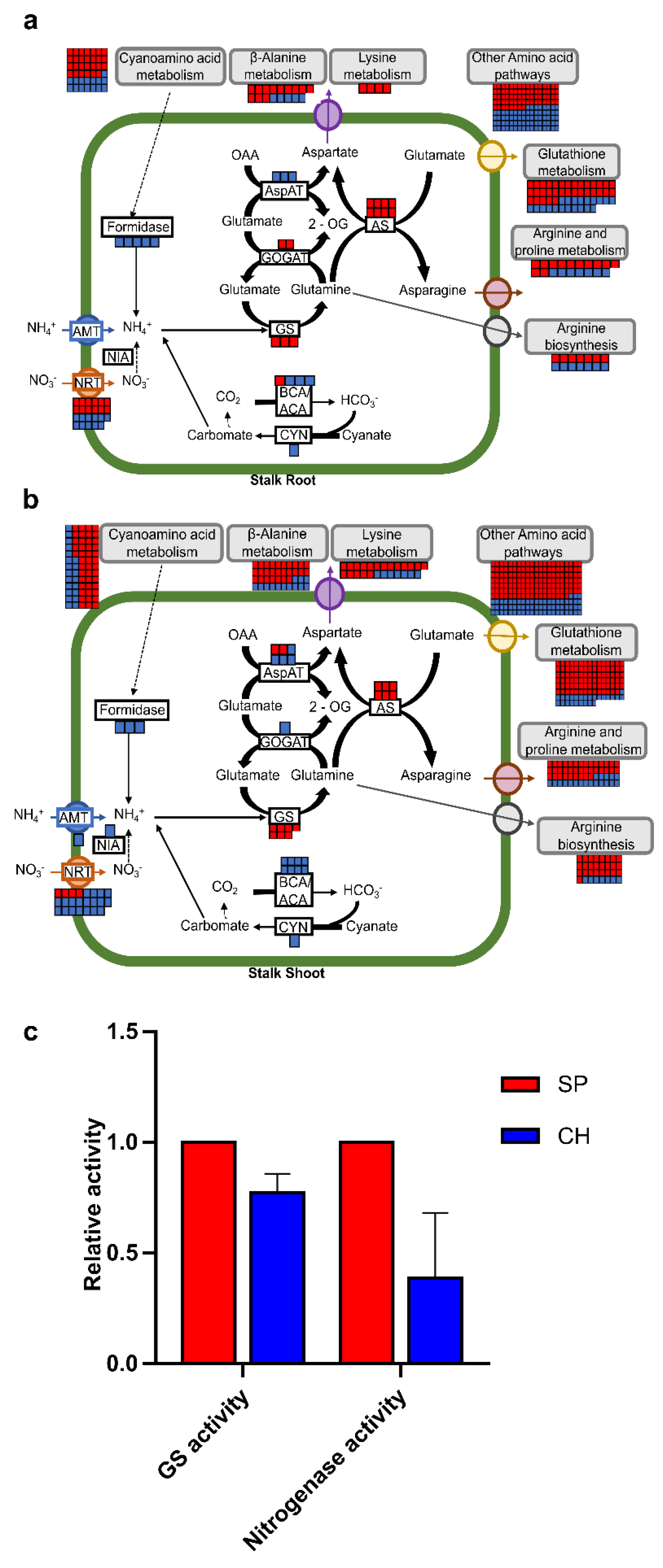
2.4. Plant N Assimilation and Metabolism and Amino Acid Metabolism Pathways
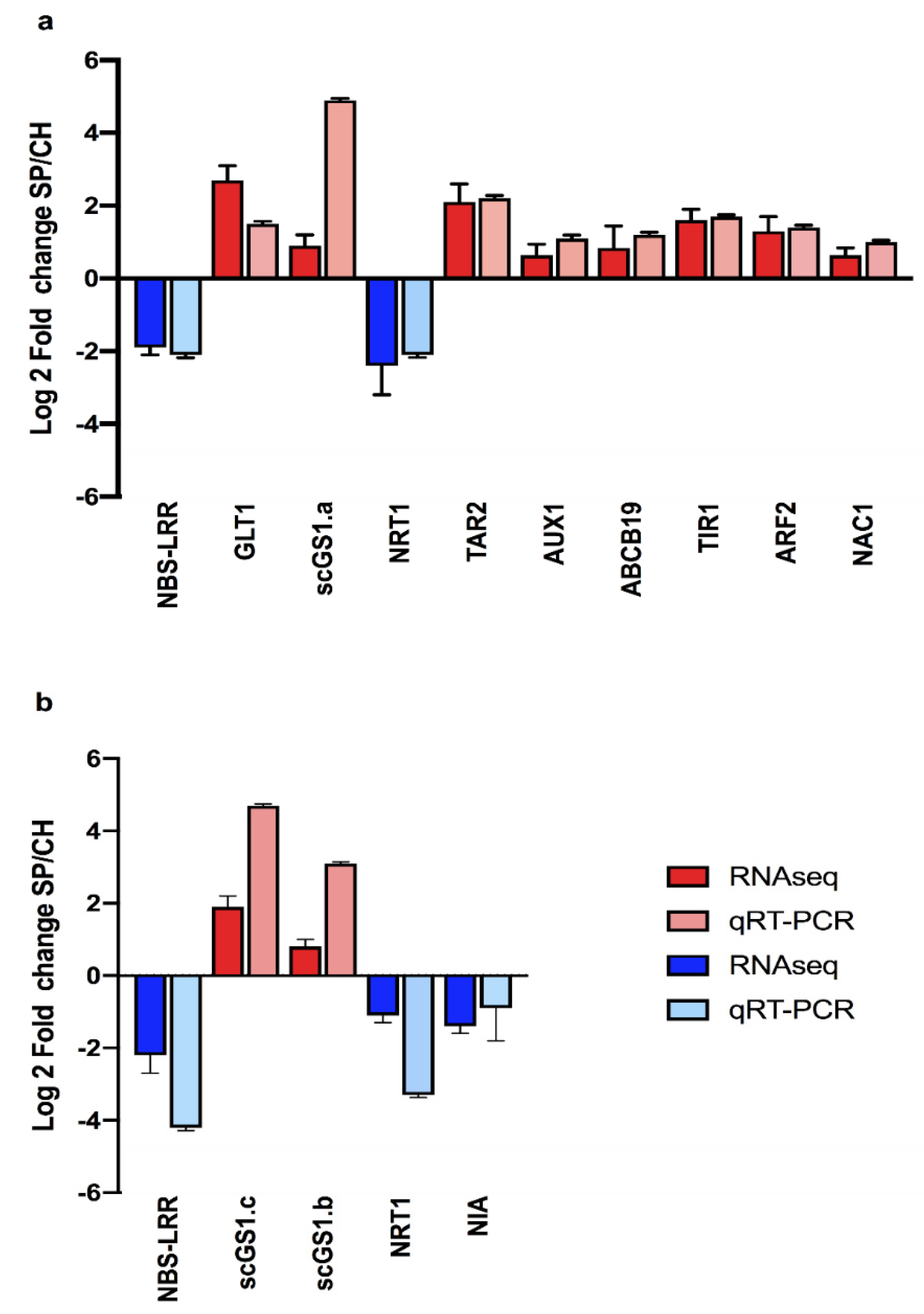
2.4.1. Nitrate Transporters
2.4.2. Amide Amino Acid Metabolic Pathway
2.4.3. Amide Amino Acid Metabolic Pathway
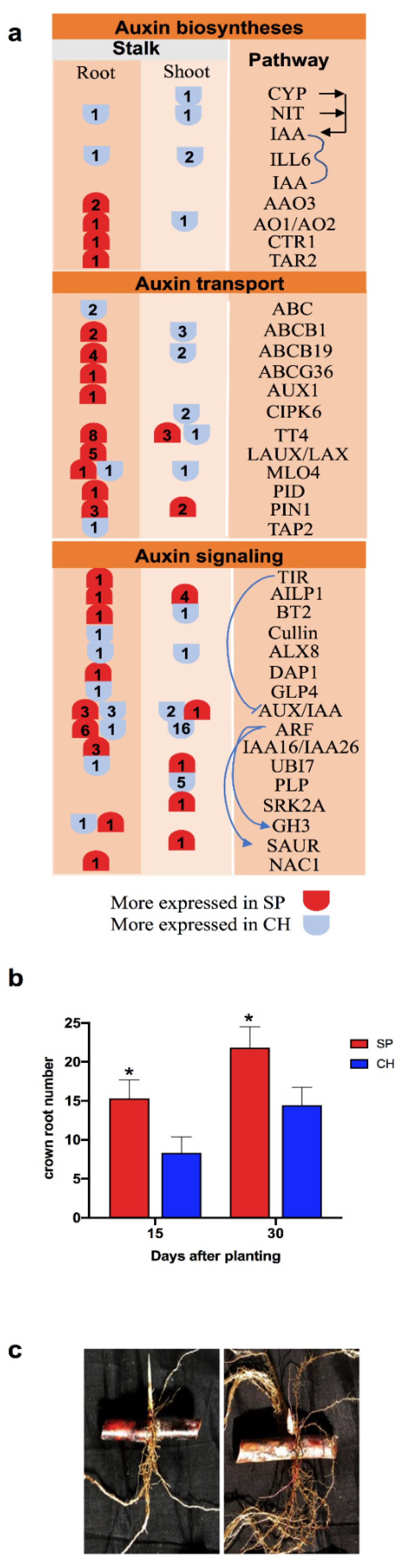
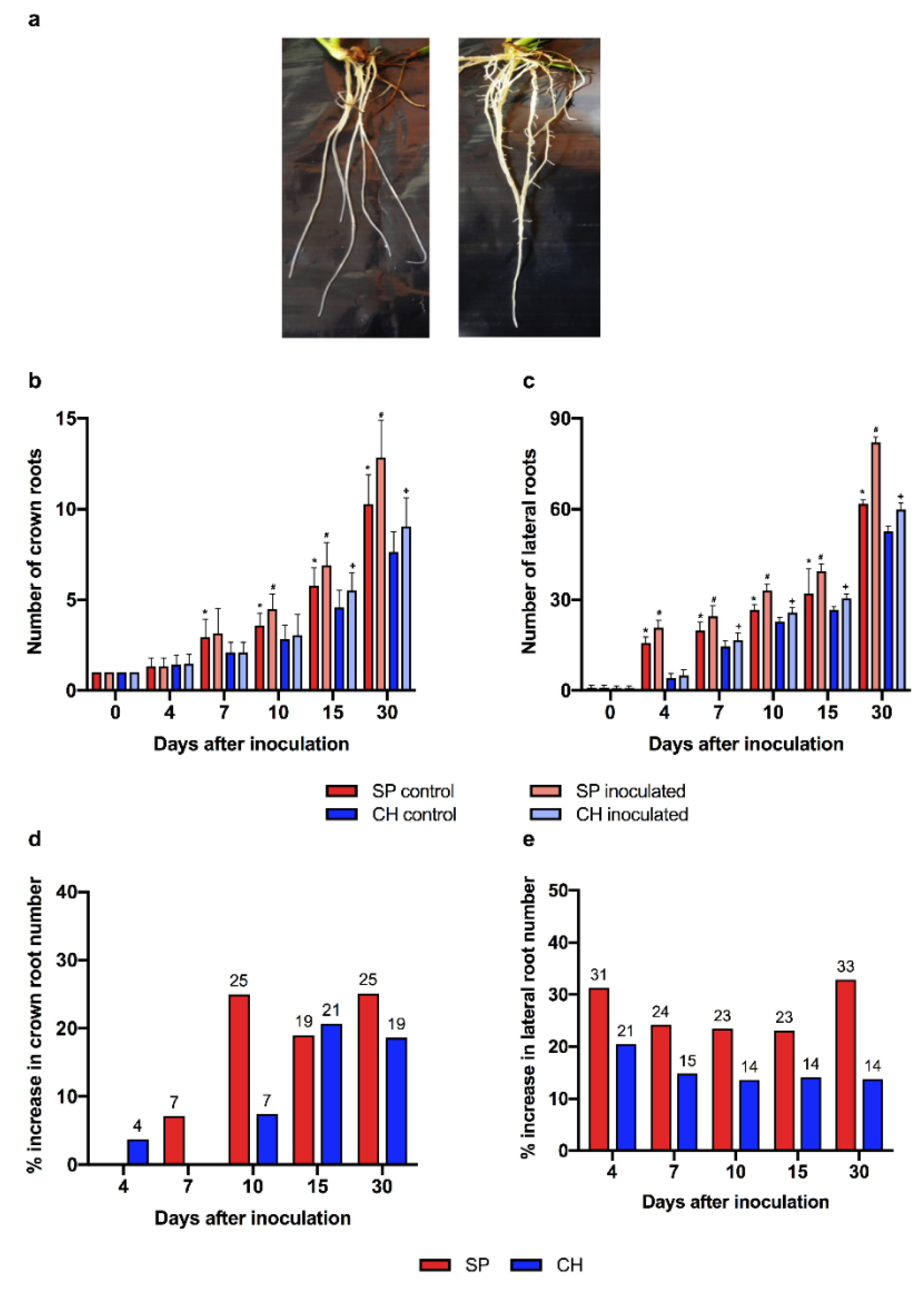
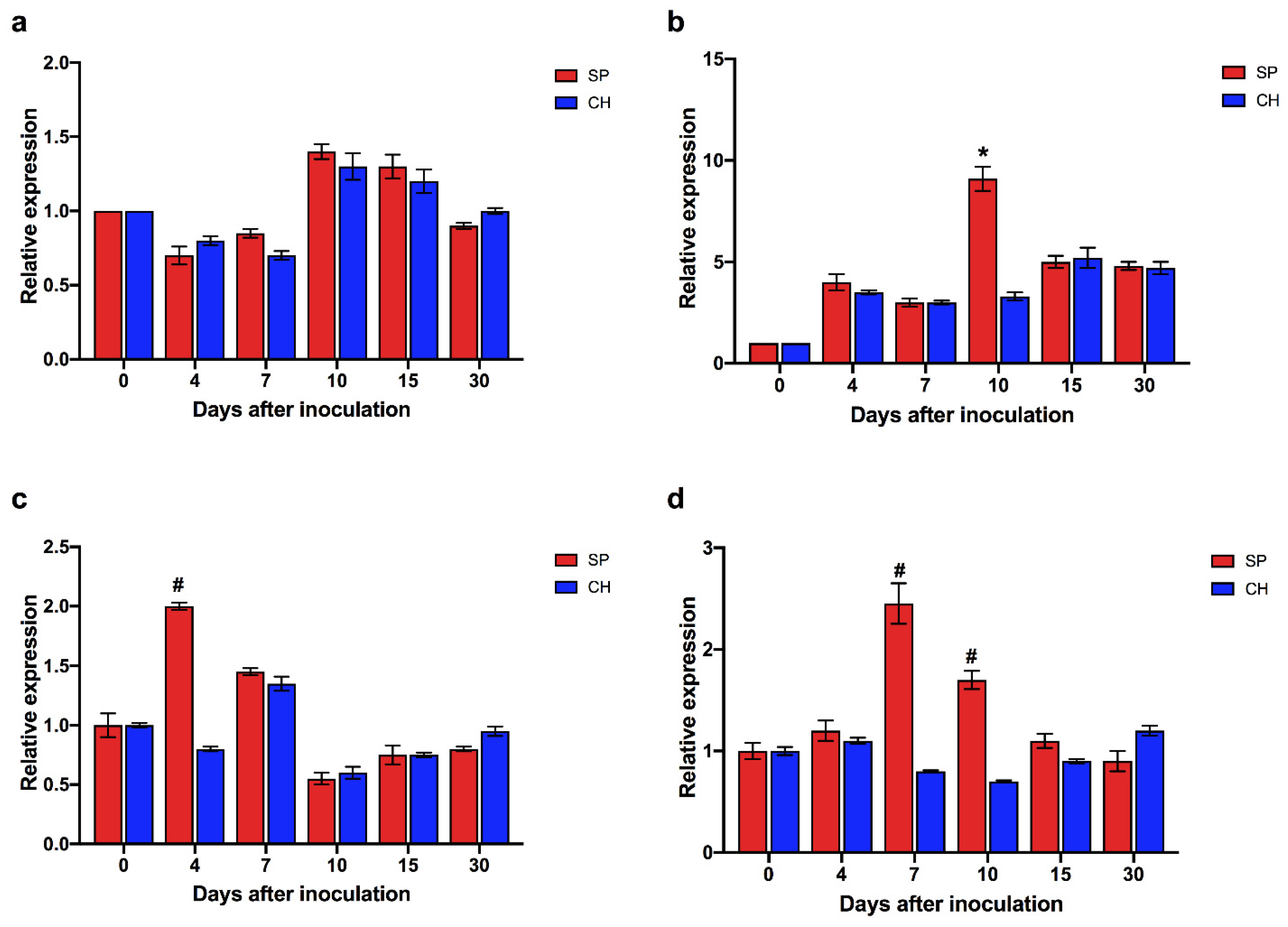
2.5. Auxin Hormonal Pathway
3. Materials and Methods
3.1. Plant Material for Library Construction and RNA Extraction
3.2. Plant Material for Experiments in Hydroponic Culture and Root Development Analysis
3.3. Bacteria Inoculation and Evaluation of Plant Colonization
3.4. Library Construction, Sequencing and De Novo Assembly
3.5. Read Mapping and Differential Expression Analysis
3.6. Functional Annotation of Sugarcane Transcripts
3.7. RNA Expression by qRT-PCR
3.8. Glutamine Synthetase and Nitrogenase Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Souza, G.M.; Berges, H.; Bocs, S.; Casu, R.; D’Hont, A.; Ferreira, J.E.; Henry, R.; Ming, R.; Potier, B.; Van Sluys, M.-A.; et al. The Sugarcane Genome Challenge: Strategies for Sequencing a Highly Complex Genome. Trop. Plant Biol. 2011, 4, 145–156. [Google Scholar] [CrossRef]
- Urquiaga, S.; Cruz, K.H.S.; Boddey, R.M. Contribution of Nitrogen Fixation to Sugar Cane: Nitrogen-15 and Nitrogen-Balance Estimates. Soil Sci. Soc. Am. J. 1992, 56, 105–114. [Google Scholar] [CrossRef]
- Urquiaga, S.; Xavier, R.P.; de Morais, R.F.; Batista, R.B.; Schultz, N.; Leite, J.M.; Maia e Sá, J.; Barbosa, K.P.; de Resende, A.S.; Alves, B.J.R.; et al. Evidence from field nitrogen balance and 15N natural abundance data for the contribution of biological N2 fixation to Brazilian sugarcane varieties. Plant Soil 2012, 356, 5–21. [Google Scholar] [CrossRef]
- Boddey, R.M.; de Oliveira, O.C.; Urquiaga, S.; Reis, V.M.; de Olivares, F.L.; Baldani, V.L.D.; Döbereiner, J. Biological nitrogen fixation associated with sugar cane and rice: Contributions and prospects for improvement. Plant Soil 1995, 174, 195–209. [Google Scholar] [CrossRef]
- Ruschel, A.P.; Vose, P.B.; Matsui, E.; Victoria, R.L.; Tsai Saito, S.M. Field evaluation of N2-fixation and N-utilization by phaseolus bean varieties determined by15N isotope dilution. Plant Soil 1982, 65, 397–407. [Google Scholar] [CrossRef]
- Boddey, R.M.; Urquiaga, S.; Alves, B.J.R.; Reis, V. Endophytic nitrogen fixation in sugarcane: Present knowledge and future applications. Plant Soil 2003, 252, 139–149. [Google Scholar] [CrossRef]
- Baldani, J.I. A brief story of nitrogen fixation in sugarcane—reasons for success in Brazil. Funct. Plant Biol. 2002, 29, 417–423. [Google Scholar] [CrossRef]
- Martins, D.S.; Reis, V.M.; Schultz, N.; Alves, B.J.R.; Urquiaga, S.; Pereira, W.; Sousa, J.S.; Boddey, R.M. Both the contribution of soil nitrogen and of biological N2 fixation to sugarcane can increase with the inoculation of diazotrophic bacteria. Plant Soil 2020, 454, 155–169. [Google Scholar] [CrossRef]
- Antunes, J.E.L.; Freitas, A.D.S.; Oliveira, L.M.S.; Lyra, M.; Fonseca, M.A.C.; Santos, C.; Oliveira, J.P.; Araújo, A.S.F.; Figueiredo, M.V.B. Sugarcane inoculated with endophytic diazotrophic bacteria: Effects on yield, biological nitrogen fixation and industrial characteristics. An. Da Acad. Bras. Cienc. 2019, 91, e20180990. [Google Scholar] [CrossRef]
- Robinson, N.; Brackin, R.; Vinall, K.; Soper, F.; Holst, J.; Gamage, H.; Paungfoo-Lonhienne, C.; Rennenberg, H.; Lakshmanan, P.; Schmidt, S. Nitrate Paradigm Does Not Hold Up for Sugarcane. PLoS ONE 2011, 6, e19045. [Google Scholar] [CrossRef] [PubMed]
- Barnett, M.J.; Toman, C.J.; Fisher, R.F.; Long, S.R. A dual-genome Symbiosis Chip for coordinate study of signal exchange and development in a prokaryote–host interaction. Proc. Natl. Acad. Sci. USA 2004, 101, 16636. [Google Scholar] [CrossRef] [PubMed]
- Baldani, J.; Caruso, L.; Baldani, V.L.D.; Goi, S.R.; Döbereiner, J. Recent advances in BNF with non-legume plants. Soil Biol. Biochem. 1997, 29, 911–922. [Google Scholar] [CrossRef]
- Carvalho, T.L.G.; Ferreira, P.C.G.; Hemerly, A.S. Sugarcane Genetic Controls Involved in the Association with Beneficial Endophytic Nitrogen Fixing Bacteria. Trop. Plant Biol. 2011, 4, 31–41. [Google Scholar] [CrossRef]
- Reinhold-Hurek, B.; Hurek, T. Life in grasses: Diazotrophic endophytes. Trends Microbiol. 1998, 6, 139–144. [Google Scholar] [CrossRef]
- Fuentes-Ramirez, L.E.; Jimenez-Salgado, T.; Abarca-Ocampo, I.R.; Caballero-Mellado, J. Acetobacter diazotrophicus, an indoleacetic acid producing bacterium isolated from sugarcane cultivars of México. Plant Soil 1993, 154, 145–150. [Google Scholar] [CrossRef]
- Oliveira, A.L.M.; Urquiaga, S.; Döbereiner, J.; Baldani, J.I. The effect of inoculating endophytic N2-fixing bacteria on micropropagated sugarcane plants. Plant Soil 2002, 242, 205–215. [Google Scholar] [CrossRef]
- Boddey, R.M.; Dobereiner, J. Nitrogen fixation associated with grasses and cereals: Recent progress and perspectives for the future. Fertil. Res. 1995, 42, 241–250. [Google Scholar] [CrossRef]
- Cavalcante, J.J.V.; Vargas, C.; Nogueira, E.M.; Vinagre, F.; Schwarcz, K.; Baldani, J.I.; Ferreira, P.C.G.; Hemerly, A.S. Members of the ethylene signalling pathway are regulated in sugarcane during the association with nitrogen-fixing endophytic bacteria. J. Exp. Bot. 2007, 58, 673–686. [Google Scholar] [CrossRef]
- Dos Santos, M.F.; Muniz de Pádua, V.L.; de Matos Nogueira, E.; Hemerly, A.S.; Domont, G.B. Proteome of Gluconacetobacter diazotrophicus co-cultivated with sugarcane plantlets. J. Proteom. 2010, 73, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, E.d.M.; Olivares, F.L.; Japiassu, J.C.; Vilar, C.; Vinagre, F.; Baldani, J.I.; Silva Hemerly, A. Characterization of glutamine synthetase genes in sugarcane genotypes with different rates of biological nitrogen fixation. Plant Sci. 2005, 169, 819–832. [Google Scholar] [CrossRef]
- Vargas, L.; Santa Brígida, A.B.; Mota Filho, J.P.; de Carvalho, T.G.; Rojas, C.A.; Vaneechoutte, D.; Van Bel, M.; Farrinelli, L.; Ferreira, P.C.G.; Vandepoele, K.; et al. Drought Tolerance Conferred to Sugarcane by Association with Gluconacetobacter diazotrophicus: A Transcriptomic View of Hormone Pathways. PLoS ONE 2014, 9, e114744. [Google Scholar] [CrossRef] [PubMed]
- Vinagre, F.; Vargas, C.; Schwarcz, K.; Cavalcante, J.; Nogueira, E.M.; Baldani, J.I.; Ferreira, P.C.; Hemerly, A.S. SHR5: A novel plant receptor kinase involved in plant-N2-fixing endophytic bacteria association. J. Exp. Bot. 2006, 57, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, H.G.F.; Rosman, A.C.; Carvalho, T.L.G.; Grativol, C.; Hemerly, A.S. Cell wall formation pathways are differentially regulated in sugarcane contrasting genotypes associated with endophytic diazotrophic bacteria. Planta 2021, 254, 109. [Google Scholar] [CrossRef] [PubMed]
- Compant, S.; Clément, C.; Sessitsch, A. Plant growth-promoting bacteria in the rhizo- and endosphere of plants: Their role, colonization, mechanisms involved and prospects for utilization. Soil Biol. Biochem. 2010, 42, 669–678. [Google Scholar] [CrossRef]
- James, E.K.; Olivares, F.L. Infection and Colonization of Sugar Cane and Other Graminaceous Plants by Endophytic Diazotrophs. Crit. Rev. Plant Sci. 1998, 17, 77–119. [Google Scholar] [CrossRef]
- James, E.K.; Olivares, F.L.; de Oliveira, A.L.M.; dos Reis, J.F.B.; da Silva, L.G.; Reis, V.M. Further observations on the interaction between sugar cane and Gluconacetobacter diazotrophicus under laboratory and greenhouse conditions1. J. Exp. Bot. 2001, 52, 747–760. [Google Scholar] [CrossRef]
- Rosenblueth, M.; Martínez-Romero, E. Bacterial Endophytes and Their Interactions with Hosts. Mol. Plant-Microbe Interact. 2006, 19, 827–837. [Google Scholar] [CrossRef]
- Ringli, C. Monitoring the Outside: Cell Wall-Sensing Mechanisms. Plant Physiol. 2010, 153, 1445. [Google Scholar] [CrossRef]
- Afzal, A.J.; Wood, A.J.; Lightfoot, D.A. Plant receptor-like serine threonine kinases: Roles in signaling and plant defense. Mol. Plant-Microbe Interact. 2008, 21, 507–517. [Google Scholar] [CrossRef]
- Antolín-Llovera, M.; Ried, M.K.; Binder, A.; Parniske, M. Receptor Kinase Signaling Pathways in Plant-Microbe Interactions. Annu. Rev. Phytopathol. 2011, 50, 451–473. [Google Scholar] [CrossRef] [PubMed]
- DeYoung, B.; Innes, R.; DeYoung, B.J.; Innes, R.W. Plant NBS-LRR proteins in pathogen sensing and host defense. Nat. Immunol. 2007, 7, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, E.d.M.; Vinagre, F.; Masuda, H.P.; Vargas, C.; Pádua, V.L.M.d.; Silva, F.R.d.; Santos, R.V.d.; Baldani, J.I.; Ferreira, P.C.G.; Hemerly, A.S. Expression of sugarcane genes induced by inoculation with Gluconacetobacter diazotrophicus and Herbaspirillum rubrisubalbicans. Genet. Mol. Biol. 2001, 24, 199–206. [Google Scholar] [CrossRef]
- Chinchilla, D.; Zipfel, C.; Robatzek, S.; Kemmerling, B.; Nürnberger, T.; Jones, J.D.; Felix, G.; Boller, T. A flagellin-induced complex of the receptor FLS2 and BAK1 initiates plant defence. Nature 2007, 448, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Trdá, L.; Fernandez, O.; Boutrot, F.; Héloir, M.C.; Kelloniemi, J.; Daire, X.; Adrian, M.; Clément, C.; Zipfel, C.; Dorey, S.; et al. The grapevine flagellin receptor VvFLS2 differentially recognizes flagellin-derived epitopes from the endophytic growth-promoting bacterium Burkholderia phytofirmans and plant pathogenic bacteria. New Phytol. 2014, 201, 1371–1384. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.E.; Klepac-Ceraj, V.; Acinas, S.G.; Gautier, C.; Bertilsson, S.; Polz, M.F. Evaluation of 23S rRNA PCR Primers for Use in Phylogenetic Studies of Bacterial Diversity. Appl. Environ. Microbiol. 2006, 72, 2221. [Google Scholar] [CrossRef]
- Matsuda, K.; Tsuji, H.; Asahara, T.; Kado, Y.; Nomoto, K. Sensitive Quantitative Detection of Commensal Bacteria by rRNA-Targeted Reverse Transcription-PCR. Appl. Environ. Microbiol. 2007, 73, 32. [Google Scholar] [CrossRef]
- Matsuda, K.; Tsuji, H.; Asahara, T.; Takahashi, T.; Kubota, H.; Nagata, S.; Yamashiro, Y.; Nomoto, K. Sensitive quantification of Clostridium difficile cells by reverse transcription-quantitative PCR targeting rRNA molecules. Appl. Environ. Microbiol. 2012, 78, 5111–5118. [Google Scholar] [CrossRef]
- Pei, A.; Nossa, C.W.; Chokshi, P.; Blaser, M.J.; Yang, L.; Rosmarin, D.M.; Pei, Z. Diversity of 23S rRNA genes within individual prokaryotic genomes. PLoS ONE 2009, 4, e5437. [Google Scholar] [CrossRef]
- Carvalho, T.L.G.; Balsemão-Pires, E.; Saraiva, R.M.; Ferreira, P.C.G.; Hemerly, A.S. Nitrogen signalling in plant interactions with associative and endophytic diazotrophic bacteria. J. Exp. Bot. 2014, 65, 5631–5642. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Cheng, Y.-H.; Chen, K.-E.; Tsay, Y.-F. Nitrate Transport, Signaling, and Use Efficiency. Annu. Rev. Plant Biol. 2018, 69, 85–122. [Google Scholar] [CrossRef]
- Xu, G.; Fan, X.; Miller, A.J. Plant nitrogen assimilation and use efficiency. Annu. Rev. Plant Biol. 2012, 63, 153–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y.; Zhu, F.; Ming, R.; Chen, L.-Q. Genome-Wide Analysis of Nitrate Transporter (NRT/NPF) Family in Sugarcane Saccharum spontaneum L. Trop. Plant Biol. 2019, 12, 133–149. [Google Scholar] [CrossRef]
- Leran, S.; Varala, K.; Boyer, J.C.; Chiurazzi, M.; Crawford, N.; Daniel-Vedele, F.; David, L.; Dickstein, R.; Fernandez, E.; Forde, B.; et al. A unified nomenclature of NITRATE TRANSPORTER 1/PEPTIDE TRANSPORTER family members in plants. Trends Plant Sci. 2014, 19, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Komarova, N.Y.; Thor, K.; Gubler, A.; Meier, S.; Dietrich, D.; Weichert, A.; Suter Grotemeyer, M.; Tegeder, M.; Rentsch, D. AtPTR1 and AtPTR5 Transport Dipeptides in Planta. Plant Physiol. 2008, 148, 856. [Google Scholar] [CrossRef] [PubMed]
- Nour-Eldin, H.H.; Andersen, T.G.; Burow, M.; Madsen, S.R.; Jørgensen, M.E.; Olsen, C.E.; Dreyer, I.; Hedrich, R.; Geiger, D.; Halkier, B.A. NRT/PTR transporters are essential for translocation of glucosinolate defence compounds to seeds. Nature 2012, 488, 531. [Google Scholar] [CrossRef] [PubMed]
- Plett, J.M.; Wilkins, O.; Campbell, M.M.; Ralph, S.G.; Regan, S. Endogenous overexpression of Populus MYB186 increases trichome density, improves insect pest resistance, and impacts plant growth. Plant J. 2010, 64, 419–432. [Google Scholar] [CrossRef]
- Kanno, Y.; Kamiya, Y.; Seo, M. Nitrate does not compete with abscisic acid as a substrate of AtNPF4.6/NRT1.2/AIT1 in Arabidopsis. Plant Signal. Behav. 2013, 8, e26624. [Google Scholar] [CrossRef]
- Karim, S.; Lundh, D.; Holmström, K.-O.; Mandal, A.; Pirhonen, M. Structural and functional characterization of AtPTR3, a stress-induced peptide transporter of Arabidopsis. J. Mol. Model. 2005, 11, 226–236. [Google Scholar] [CrossRef]
- Chiba, Y.; Shimizu, T.; Miyakawa, S.; Kanno, Y.; Koshiba, T.; Kamiya, Y.; Seo, M. Identification of Arabidopsis thaliana NRT1/PTR FAMILY (NPF) proteins capable of transporting plant hormones. J. Plant Res. 2015, 128, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Kuo, H.F.; Canivenc, G.; Lin, C.S.; Lepetit, M.; Hsu, P.K.; Tillard, P.; Lin, H.L.; Wang, Y.Y.; Tsai, C.B.; et al. Mutation of the Arabidopsis NRT1.5 nitrate transporter causes defective root-to-shoot nitrate transport. Plant Cell 2008, 20, 2514–2528. [Google Scholar] [CrossRef]
- Campbell, W.H. Nitrate reductase structure, function and regulation: Bridging the Gap between Biochemistry and Physiology. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999, 50, 277–303. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Stöhr, C. Soluble and Plasma Membrane-bound Enzymes Involved in Nitrate and Nitrite Metabolism. In Photosynthetic Nitrogen Assimilation and Associated Carbon and Respiratory Metabolism; Foyer, C.H., Noctor, G., Eds.; Springer: Dordrecht, The Netherlands, 2002; pp. 49–62. [Google Scholar]
- Zhang, J.; Zhang, X.; Tang, H.; Zhang, Q.; Hua, X.; Ma, X.; Zhu, F.; Jones, T.; Zhu, X.; Bowers, J.; et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.; Holmström, K.-O.; Mandal, A.; Dahl, P.; Hohmann, S.; Brader, G.; Palva, E.T.; Pirhonen, M. AtPTR3, a wound-induced peptide transporter needed for defence against virulent bacterial pathogens in Arabidopsis. Planta 2007, 225, 1431–1445. [Google Scholar] [CrossRef]
- Léran, S.; Noguero, M.; Corratgé-Faillie, C.; Boursiac, Y.; Brachet, C.; Lacombe, B. Functional Characterization of the Arabidopsis Abscisic Acid Transporters NPF4.5 and NPF4.6 in Xenopus Oocytes. Front. Plant Sci. 2020, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Bharath, P.; Gahir, S.; Raghavendra, A.S. Abscisic Acid-Induced Stomatal Closure: An Important Component of Plant Defense Against Abiotic and Biotic Stress. Front. Plant Sci. 2021, 12, 615114. [Google Scholar] [CrossRef] [PubMed]
- Babst, B.A.; Gao, F.; Acosta-Gamboa, L.M.; Karve, A.; Schueller, M.J.; Lorence, A. Three NPF genes in Arabidopsis are necessary for normal nitrogen cycling under low nitrogen stress. Plant Physiol. Biochem. 2019, 143, 1–10. [Google Scholar] [CrossRef]
- Chen, C.-Z.; Lv, X.-F.; Li, J.-Y.; Yi, H.-Y.; Gong, J.-M. Arabidopsis NRT1.5 is Another Essential Component in the Regulation of Nitrate Reallocation and Stress Tolerance. Plant Physiol. 2012, 159, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Obulareddy, N.; Panchal, S.; Melotto, M. Guard Cell Purification and RNA Isolation Suitable for High-Throughput Transcriptional Analysis of Cell-Type Responses to Biotic Stresses. Mol. Plant-Microbe Interact. 2013, 26, 844–849. [Google Scholar] [CrossRef]
- Masclaux-Daubresse, C.; Daniel-Vedele, F.; Dechorgnat, J.; Chardon, F.; Gaufichon, L.; Suzuki, A. Nitrogen uptake, assimilation and remobilization in plants: Challenges for sustainable and productive agriculture. Ann. Bot. 2010, 105, 1141–1157. [Google Scholar] [CrossRef]
- Marcos, F.C.C.; Iório, R.d.P.F.; Silveira, A.P.D.d.; Ribeiro, R.V.; Machado, E.C.; Lagôa, A.M.M.d.A. Endophytic bacteria affect sugarcane physiology without changing plant growth. Bragantia 2016, 75, 1–9. [Google Scholar] [CrossRef]
- Aguiar, N.; Olivares, F.; Novotny, E.; Canellas, L. Changes in metabolic profiling of sugarcane leaves induced by endophytic diazotrophic bacteria and humic acids. PeerJ 2018, 6, e5445. [Google Scholar] [CrossRef] [PubMed]
- Elisabeth, P.; Anis, M.L. Amino acid synthesis under abiotic stress. In Amino Acids in Higher Plants; D’Mello, J.P.F., Ed.; CAB International: Wallingford, UK, 2015; pp. 262–276. [Google Scholar]
- Yang, Q.; Zhao, D.; Liu, Q. Connections Between Amino Acid Metabolisms in Plants: Lysine as an Example. Front. Plant Sci. 2020, 11, 928. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, T.M.; Nunes Nesi, A.; Araújo, W.L.; Braun, H.P. Amino Acid Catabolism in Plants. Mol. Plant 2015, 8, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Obata, T.; Fernie, A.R. The use of metabolomics to dissect plant responses to abiotic stresses. Cell. Mol. Life Sci. 2012, 69, 3225–3243. [Google Scholar] [CrossRef]
- Good, A.G.; Johnson, S.J.; De Pauw, M.; Carroll, R.T.; Savidov, N.; Vidmar, J.; Lu, Z.; Taylor, G.; Stroeher, V. Engineering nitrogen use efficiency with alanine aminotransferase. Can. J. Bot. 2007, 85, 252–262. [Google Scholar] [CrossRef]
- Shrawat, A.K.; Carroll, R.T.; DePauw, M.; Taylor, G.J.; Good, A.G. Genetic engineering of improved nitrogen use efficiency in rice by the tissue-specific expression of alanine aminotransferase. Plant Biotechnol. J. 2008, 6, 722–732. [Google Scholar] [CrossRef]
- Snyman, S.J.; Hajari, E.; Watt, M.P.; Lu, Y.; Kridl, J.C. Improved nitrogen use efficiency in transgenic sugarcane: Phenotypic assessment in a pot trial under low nitrogen conditions. Plant Cell Rep. 2015, 34, 667–669. [Google Scholar] [CrossRef]
- Lam, H.-M.; Wong, P.; Chan, H.-K.; Yam, K.-M.; Chen, L.; Chow, C.-M.; Coruzzi, G.M. Overexpression of the ASN1 Gene Enhances Nitrogen Status in Seeds of Arabidopsis. Plant Physiol. 2003, 132, 926. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Park, J.; Lee, J.; Shin, D.; Marmagne, A.; Lim, P.O.; Masclaux-Daubresse, C.; An, G.; Nam, H.G. OsASN1 Overexpression in Rice Increases Grain Protein Content and Yield under Nitrogen-Limiting Conditions. Plant Cell Physiol. 2020, 61, 1309–1320. [Google Scholar] [CrossRef]
- Wang, S.; Han, K.; Peng, J.; Zhao, J.; Jiang, L.; Lu, Y.; Zheng, H.; Lin, L.; Chen, J.; Yan, F. NbALD1 mediates resistance to turnip mosaic virus by regulating the accumulation of salicylic acid and the ethylene pathway in Nicotiana benthamiana. Mol. Plant Pathol. 2019, 20, 990–1004. [Google Scholar] [CrossRef]
- Ji, Y.; Li, Q.; Liu, G.; Selvaraj, G.; Zheng, Z.; Zou, J.; Wei, Y. Roles of Cytosolic Glutamine Synthetases in Arabidopsis Development and Stress Responses. Plant Cell Physiol. 2019, 60, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Konishi, N.; Saito, M.; Imagawa, F.; Kanno, K.; Yamaya, T.; Kojima, S. Cytosolic Glutamine Synthetase Isozymes Play Redundant Roles in Ammonium Assimilation Under Low-Ammonium Conditions in Roots of Arabidopsis thaliana. Plant Cell Physiol. 2018, 59, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Konishi, N.; Ishiyama, K.; Beier, M.P.; Inoue, E.; Kanno, K.; Yamaya, T.; Takahashi, H.; Kojima, S. Contributions of two cytosolic glutamine synthetase isozymes to ammonium assimilation in Arabidopsis roots. J. Exp. Bot. 2017, 68, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Loiret, F.G.; Grimm, B.; Hajirezaei, M.R.; Kleiner, D.; Ortega, E. Inoculation of sugarcane with Pantoea sp. increases amino acid contents in shoot tissues; serine, alanine, glutamine and asparagine permit concomitantly ammonium excretion and nitrogenase activity of the bacterium. J. Plant Physiol. 2009, 166, 1152–1161. [Google Scholar] [CrossRef]
- Malviya, M.K.; Li, C.-N.; Solanki, M.K.; Singh, R.K.; Htun, R.; Singh, P.; Verma, K.K.; Yang, L.-T.; Li, Y.-R. Comparative analysis of sugarcane root transcriptome in response to the plant growth-promoting Burkholderia anthina MYSP113. PLoS ONE 2020, 15, e0231206. [Google Scholar] [CrossRef]
- Bari, R.; Jones, J.D. Role of plant hormones in plant defence responses. Plant Mol. Biol. 2009, 69, 473–488. [Google Scholar] [CrossRef]
- Patten, C.L.; Glick, B.R. Bacterial biosynthesis of indole-3-acetic acid. Can. J. Microbiol. 1996, 42, 207–220. [Google Scholar] [CrossRef]
- Vessey, J. Plant growth promoting rhizobacteria as biofertilizer. Plant Soil 2003, 255, 571–586. [Google Scholar] [CrossRef]
- Korasick, D.A.; Enders, T.A.; Strader, L.C. Auxin biosynthesis and storage forms. J. Exp. Bot. 2013, 64, 2541–2555. [Google Scholar] [CrossRef]
- Lavy, M.; Estelle, M. Mechanisms of auxin signaling. Development 2016, 143, 3226–3229. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Park, J.Y.; Kim, Y.S.; Staswick, P.E.; Jeon, J.; Yun, J.; Kim, S.Y.; Kim, J.; Lee, Y.H.; Park, C.M. GH3-mediated auxin homeostasis links growth regulation with stress adaptation response in Arabidopsis. J. Biol. Chem. 2007, 282, 10036–10046. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Mou, M.; Li, P.; Huang, Y.; Zhai, X.; Ma, Y.; Liu, J.; Yu, X. Theoretical modeling of the Aux/IAA negative feedback circuit in plants. S. Afr. J. Bot. 2015, 100, 16–19. [Google Scholar] [CrossRef]
- Xie, Q.; Frugis, G.; Colgan, D.; Chua, N.H. Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Dev. 2000, 14, 3024–3036. [Google Scholar] [CrossRef]
- Li, J.; Guo, G.; Guo, W.; Guo, G.; Tong, D.; Ni, Z.; Sun, Q.; Yao, Y. miRNA164-directed cleavage of ZmNAC1 confers lateral root development in maize (Zea mays L.). BMC Plant Biol. 2012, 12, 220. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, S.; Friml, J. Auxin: A Trigger for Change in Plant Development. Cell 2009, 136, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Woodward, A.W. Auxin: Regulation, Action, and Interaction. Ann. Bot. 2005, 95, 707–735. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kurata, T.; Okada, K.; Wada, T. A genetic regulatory network in the development of trichomes and root hairs. Annu. Rev. Plant Biol. 2008, 59, 365–386. [Google Scholar] [CrossRef] [PubMed]
- Pitts, R.J.; Cernac, A.; Estelle, M. Auxin and ethylene promote root hair elongation in Arabidopsis. Plant J. 1998, 16, 553–560. [Google Scholar] [CrossRef]
- Rahman, A.; Hosokawa, S.; Oono, Y.; Amakawa, T.; Goto, N.; Tsurumi, S. Auxin and Ethylene Response Interactions during Arabidopsis Root Hair Development Dissected by Auxin Influx Modulators. Plant Physiol. 2002, 130, 1908. [Google Scholar] [CrossRef]
- Ma, W.; Li, J.; Qu, B.; He, X.; Zhao, X.; Li, B.; Fu, X.; Tong, Y. Auxin biosynthetic gene TAR2 is involved in low nitrogen-mediated reprogramming of root architecture in Arabidopsis. Plant J. Cell Mol. Biol. 2014, 78, 70–79. [Google Scholar] [CrossRef]
- Xu, M.; Zhu, L.; Shou, H.; Wu, P. A PIN1 Family Gene, OsPIN1, involved in Auxin-dependent Adventitious Root Emergence and Tillering in Rice. Plant Cell Physiol. 2005, 46, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Marchant, A.; Bhalerao, R.; Casimiro, I.; Eklöf, J.; Casero, P.J.; Bennett, M.; Sandberg, G. AUX1 promotes lateral root formation by facilitating indole-3-acetic acid distribution between sink and source tissues in the Arabidopsis seedling. Plant Cell 2002, 14, 589–597. [Google Scholar] [CrossRef]
- Nibau, C.; Gibbs, D.J.; Coates, J.C. Branching out in new directions: The control of root architecture by lateral root formation. New Phytol. 2008, 179, 595–614. [Google Scholar] [CrossRef]
- Marin, E.; Jouannet, V.; Herz, A.; Lokerse, A.S.; Weijers, D.; Vaucheret, H.; Nussaume, L.; Crespi, M.D.; Maizel, A. miR390, Arabidopsis TAS3 tasiRNAs, and Their AUXIN RESPONSE FACTOR Targets Define an Autoregulatory Network Quantitatively Regulating Lateral Root Growth. Plant Cell 2010, 22, 1104. [Google Scholar] [CrossRef] [PubMed]
- Arencibia, A.D.; Vinagre, F.; Estevez, Y.; Bernal, A.; Perez, J.; Cavalcanti, J.; Santana, I.; Hemerly, A.S. Gluconoacetobacter diazotrophicus Elicitate a Sugarcane Defense Response Against a Pathogenic Bacteria Xanthomonas albilineans. Plant Signal. Behav. 2006, 1, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Spaepen, S.; Dobbelaere, S.; Croonenborghs, A.; Vanderleyden, J. Effects of Azospirillum brasilense indole-3-acetic acid production on inoculated wheat plants. Plant Soil 2008, 312, 15–23. [Google Scholar] [CrossRef]
- Nuruzzaman, M.; Sharoni, A.M.; Kikuchi, S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 2013, 4, 248. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Ying, S.; Zhang, D.-F.; Shi, Y.-S.; Song, Y.-C.; Wang, T.-Y.; Li, Y. A maize stress-responsive NAC transcription factor, ZmSNAC1, confers enhanced tolerance to dehydration in transgenic Arabidopsis. Plant Cell Rep. 2012, 31, 1701–1711. [Google Scholar] [CrossRef]
- Hu, H.; Dai, M.; Yao, J.; Xiao, B.; Li, X.; Zhang, Q.; Xiong, L. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 12987. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, D.R.; Arnon, D.I. The water-culture method for growing plants without soil. Circular. Calif. Agric. Exp. Stn. 1950, 347, 32. [Google Scholar]
- Logemann, J.; Schell, J.; Willmitzer, L. Improved method for the isolation of RNA from plant tissues. Anal. Biochem. 1987, 163, 16–20. [Google Scholar] [CrossRef]
- Zerbino, D.R. Using the Velvet de novo assembler for short-read sequencing technologies. Curr. Protoc. Bioinform. 2010, 1, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruan, J.; Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008, 18, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Liu, X.; Peng, Z.; Wan, Y.; Ji, Y.; He, W.; Wan, W.; Luo, J.; Guo, H. AHD2.0: An update version of Arabidopsis Hormone Database for plant systematic studies. Nucleic Acids Res. 2011, 39, D1123–D1129. [Google Scholar] [CrossRef] [PubMed]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. mapman: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.G.d.; da Silva Ribeiro, F.; Alves, G.C.; Santos, L.A.; Reis, V.M. Inoculation with five diazotrophs alters nitrogen metabolism during the initial growth of sugarcane varieties with contrasting responses to added nitrogen. Plant Soil 2019, 451, 25–44. [Google Scholar] [CrossRef]
- Medeiros, A.F.A.; Polidoro, J.C.; Reis, V.M. Nitrogen Source Effect on Gluconacetobacter diazotrophicus Colonization of Sugarcane (Saccharum spp.). Plant Soil 2006, 279, 141–152. [Google Scholar] [CrossRef]
- Vargas, C.; Pádua, V.L.; Nogueira, E.; Vinagre, F.; Masuda, H.; da Silva, F.; Baldani, J.; Ferreira, P.; Hemerly, A. Signaling pathways mediating the association between sugarcane and endophytic diazotrophic bacteria: A genomic approach. Symbiosis 2003, 35, 159–180. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant Species | Total Proteins a | Mapped Proteins b | Sugarcane loci c | % Sugarcane loci d |
|---|---|---|---|---|
| Arabidopsis | 35,386 | 14,920 | 34,617 | 64.83 |
| Rice | 49,061 | 19,639 | 40,839 | 76.48 |
| Maize | 63,540 | 23,462 | 42,438 | 79.47 |
| Sorghum | 29,448 | 19,545 | 43,715 | 81.87 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carvalho, T.L.G.; Rosman, A.C.; Grativol, C.; de M. Nogueira, E.; Baldani, J.I.; Hemerly, A.S. Sugarcane Genotypes with Contrasting Biological Nitrogen Fixation Efficiencies Differentially Modulate Nitrogen Metabolism, Auxin Signaling, and Microorganism Perception Pathways. Plants 2022, 11, 1971. https://doi.org/10.3390/plants11151971
Carvalho TLG, Rosman AC, Grativol C, de M. Nogueira E, Baldani JI, Hemerly AS. Sugarcane Genotypes with Contrasting Biological Nitrogen Fixation Efficiencies Differentially Modulate Nitrogen Metabolism, Auxin Signaling, and Microorganism Perception Pathways. Plants. 2022; 11(15):1971. https://doi.org/10.3390/plants11151971
Chicago/Turabian StyleCarvalho, Thais Louise G., Aline C. Rosman, Clícia Grativol, Eduardo de M. Nogueira, José Ivo Baldani, and Adriana S. Hemerly. 2022. "Sugarcane Genotypes with Contrasting Biological Nitrogen Fixation Efficiencies Differentially Modulate Nitrogen Metabolism, Auxin Signaling, and Microorganism Perception Pathways" Plants 11, no. 15: 1971. https://doi.org/10.3390/plants11151971
APA StyleCarvalho, T. L. G., Rosman, A. C., Grativol, C., de M. Nogueira, E., Baldani, J. I., & Hemerly, A. S. (2022). Sugarcane Genotypes with Contrasting Biological Nitrogen Fixation Efficiencies Differentially Modulate Nitrogen Metabolism, Auxin Signaling, and Microorganism Perception Pathways. Plants, 11(15), 1971. https://doi.org/10.3390/plants11151971