
Gene Losses and Plastome Degradation in the Hemiparasitic Species Plicosepalus acaciae and Plicosepalus curviflorus: Comparative Analyses and Phylogenetic Relationships among Santalales Members


Abstract
1. Introduction
2. Results
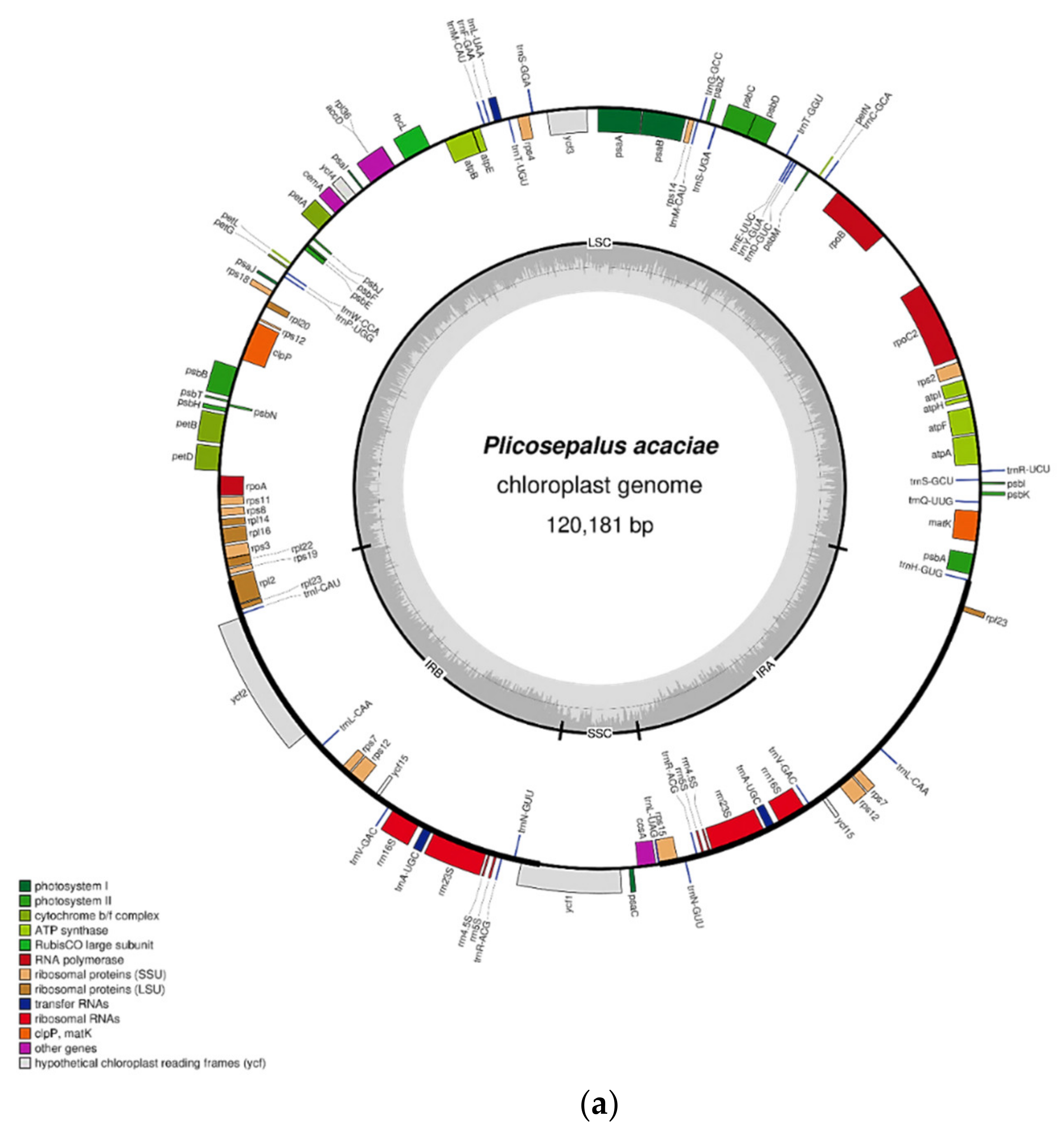
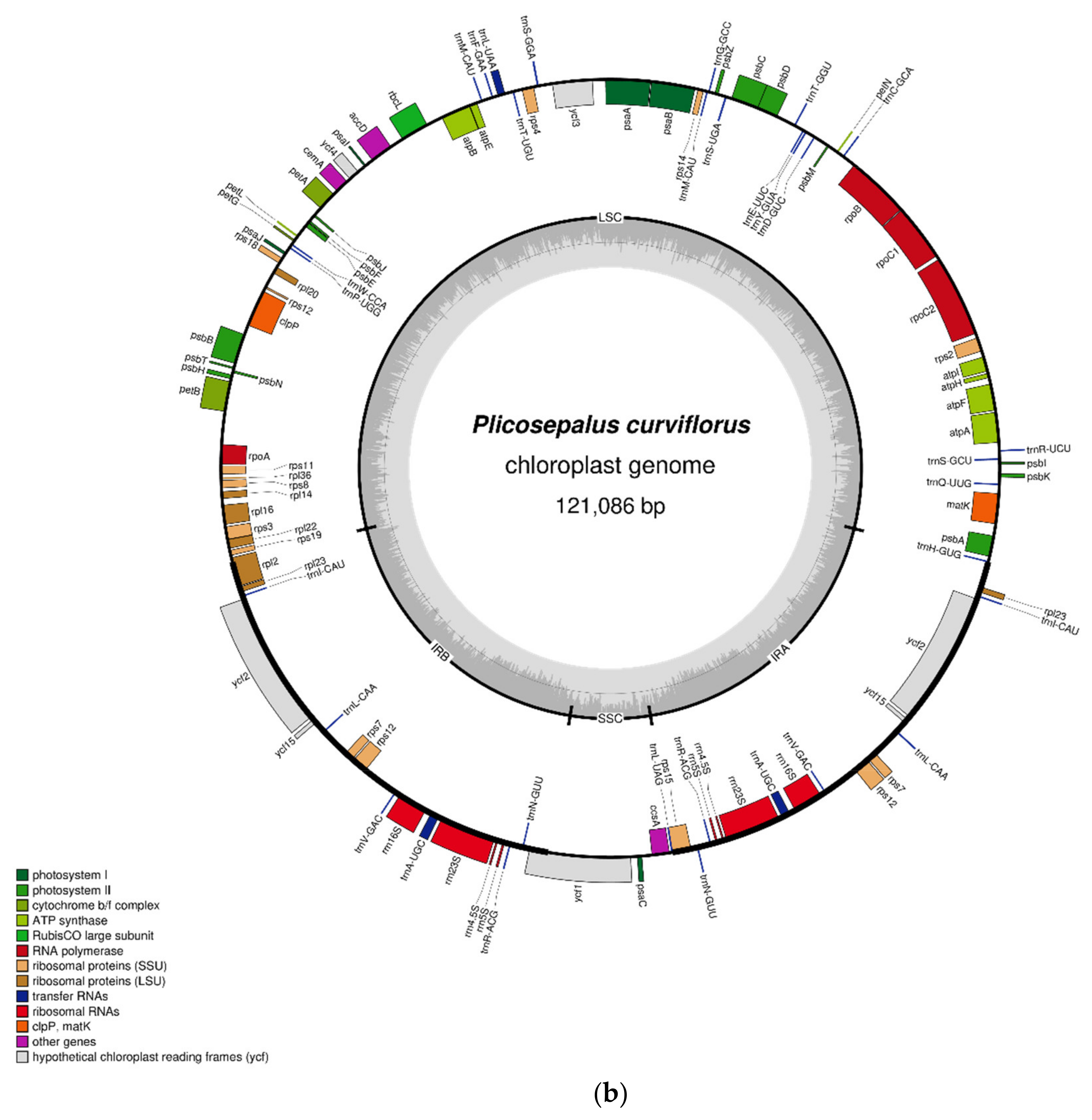
2.1. Characteristics of the Chloroplast Genome
2.2. Relative Synonymous Codon Usage (RSCU)
2.3. RNA Editing Sites
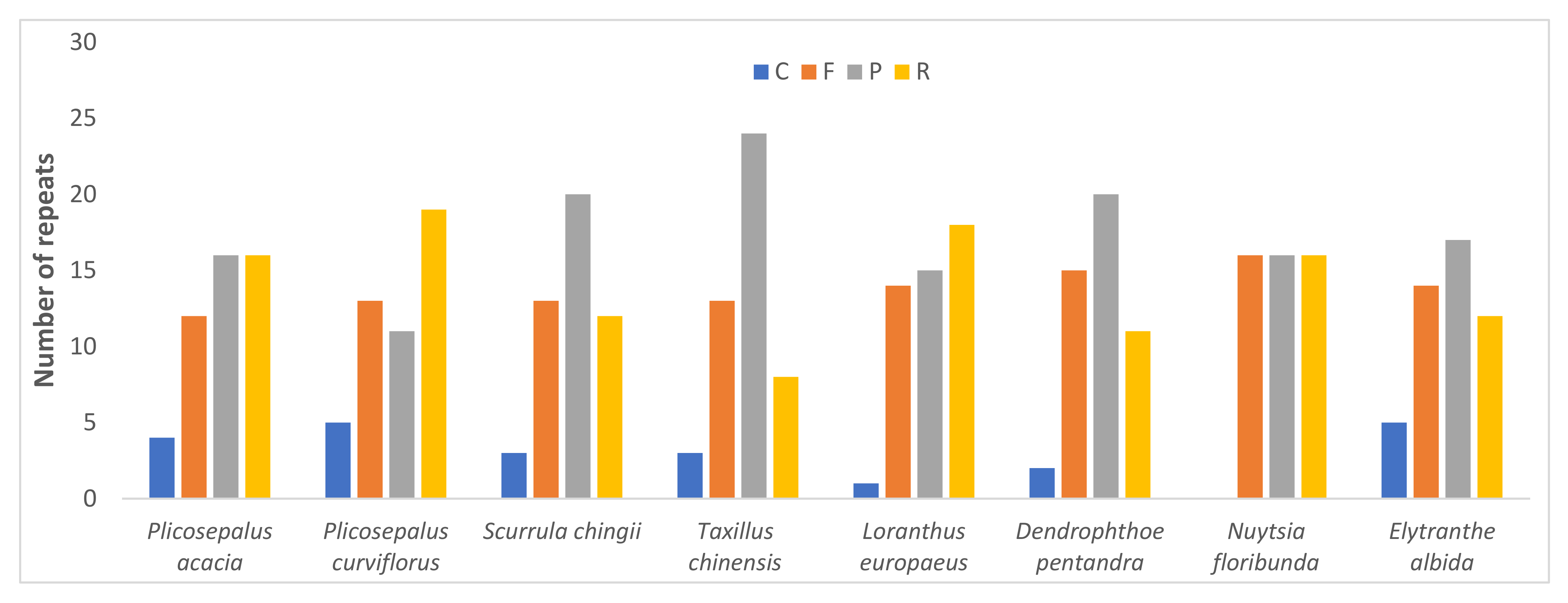
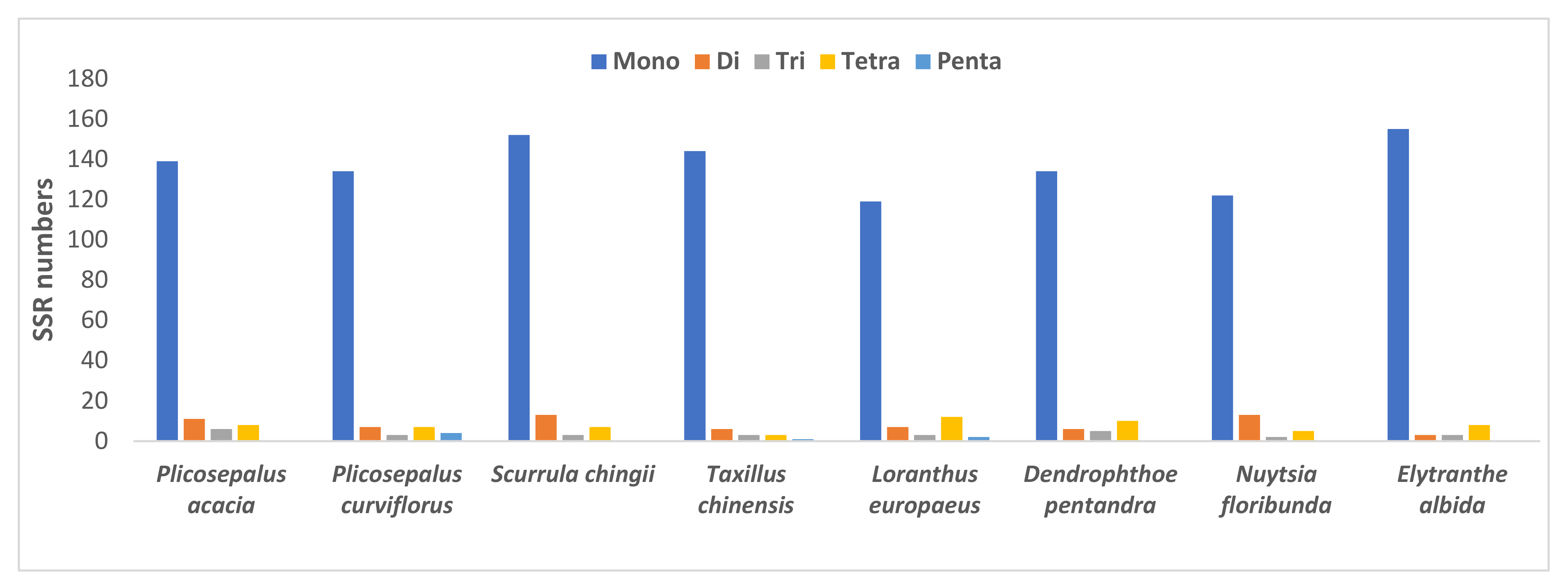
2.4. Repeat Analysis
2.4.1. Long Repeats
2.4.2. Simple Sequence Repeats
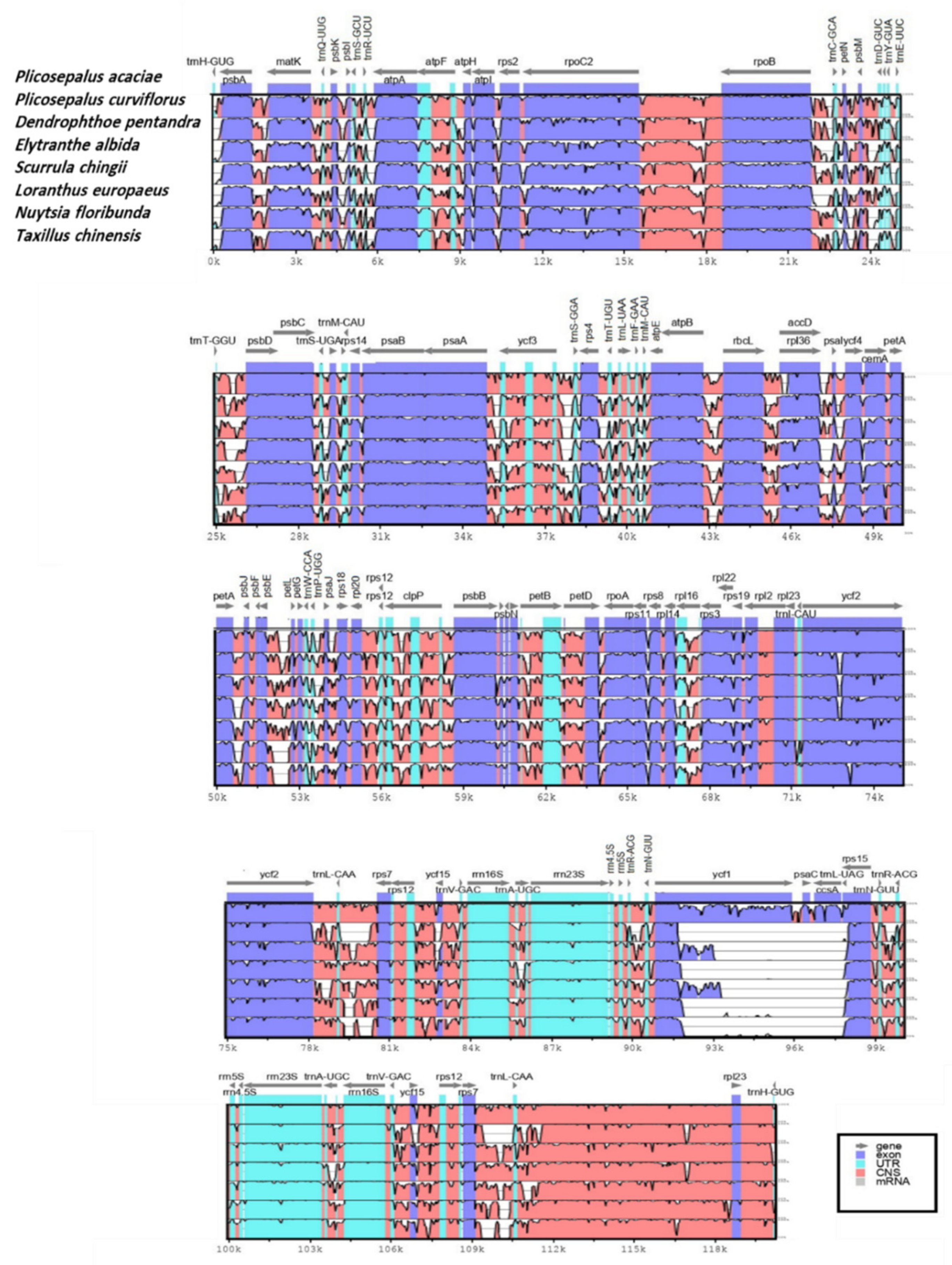
2.5. Sequence Divergence
2.6. Boundary between LSC/SSC and IRs
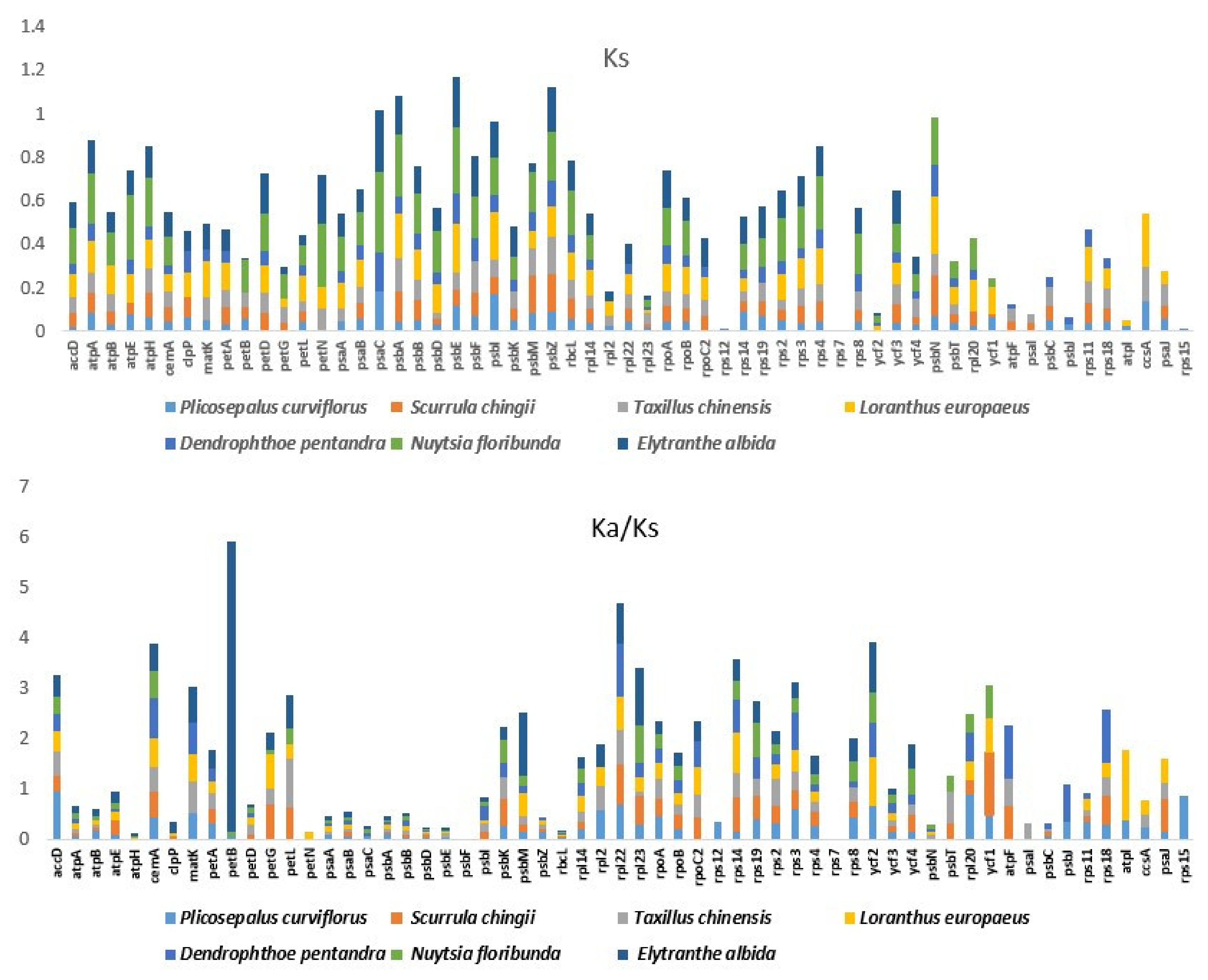
2.7. Divergence of Protein-Coding Gene Sequences
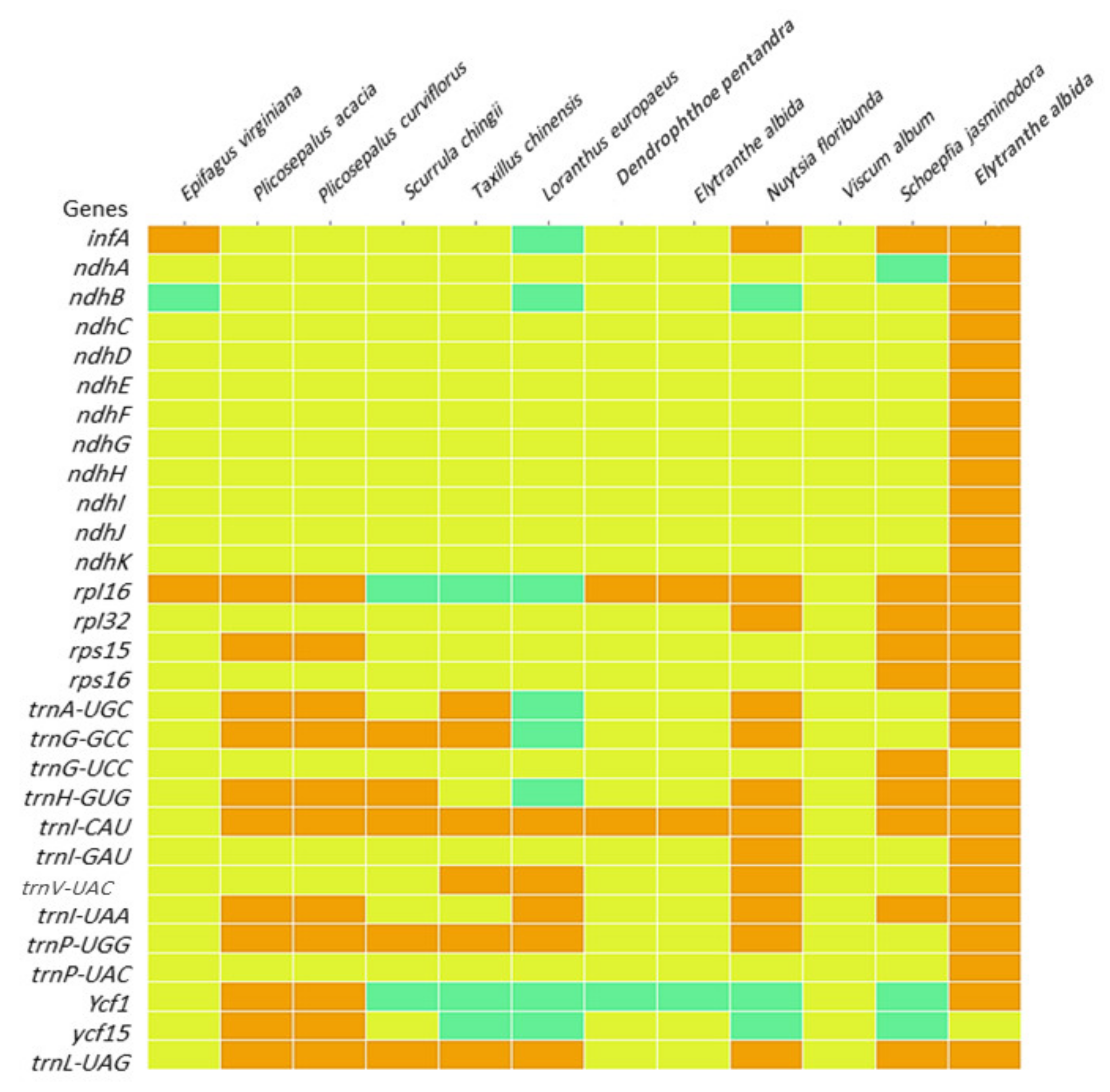
2.8. Heatmap
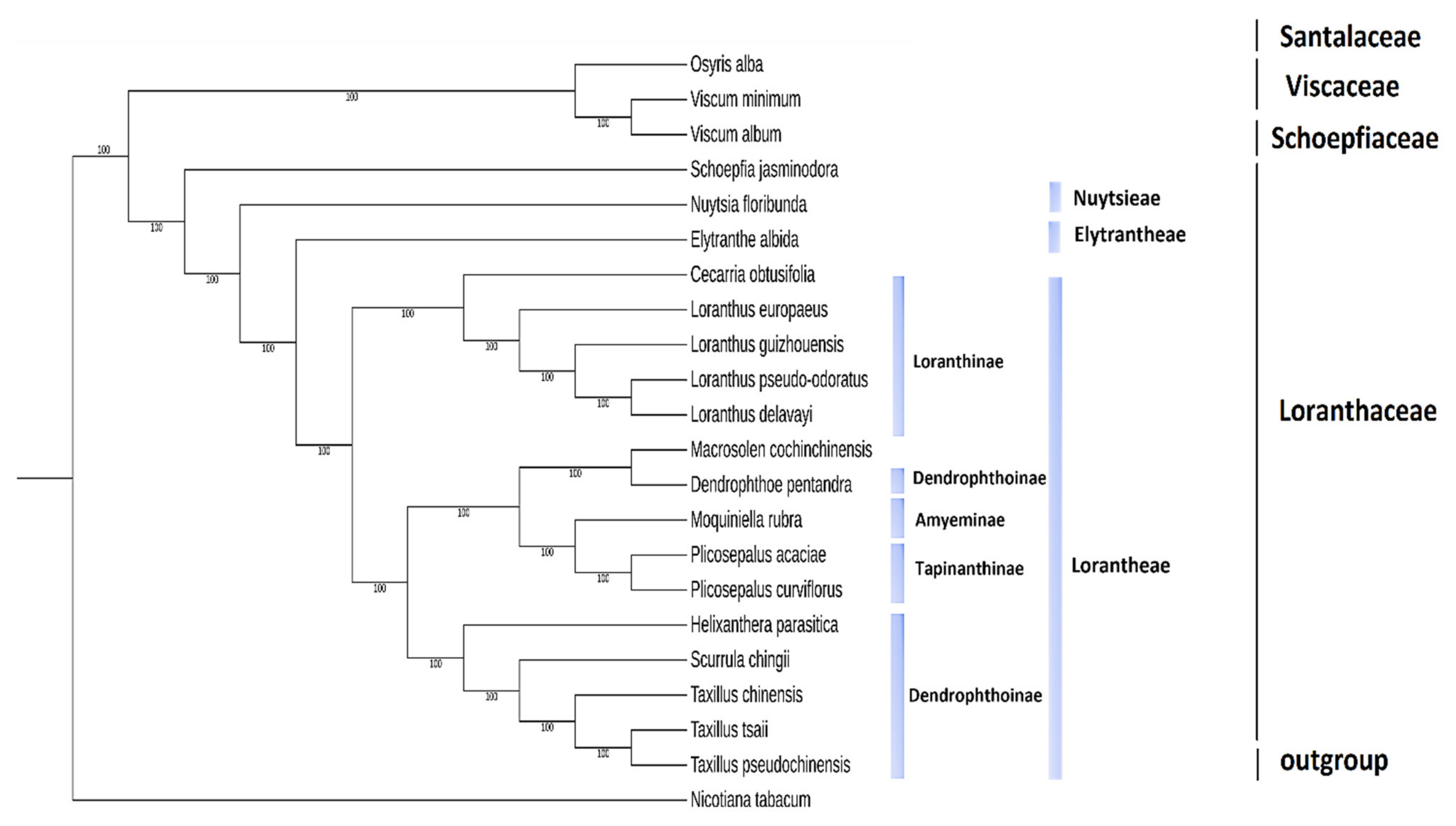
2.9. Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Library Construction
4.3. De Novo Genome Sequencing
4.4. Genome Assembly and Annotation
4.5. Sequence Analysis
4.6. Repeat Analysis in the Chloroplast Genome
4.7. Sequence Divergence and Boundary
4.8. Characterization of the Substitution Rate
4.9. Heatmap
4.10. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Shin, H.W.; Lee, N.S. Understanding plastome evolution in hemiparasitic Santalales: Complete chloroplast genomes of three species, dendrotrophe varians, helixanthera parasitica, and macrosolen cochinchinensis. PLoS ONE 2018, 13, e0200293. [Google Scholar] [CrossRef] [PubMed]
- Petersen, G.; Cuenca, A.; Seberg, O. Plastome evolution in hemiparasitic mistletoes. Genome Biol. Evol. 2015, 7, 2520–2532. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Le, C.T.; Barrett, R.L.; Nickrent, D.L.; Chen, Z.; Lu, L.; Vidal-Russell, R. Historical biogeography of Loranthaceae (Santalales): Diversification agrees with emergence of tropical forests and radiation of songbirds. Mol. Phylogenet. Evol. 2018, 124, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Su, H.-J.; Liang, S.-l.; Nickrent, D.L. Plastome variation and phylogeny of Taxillus (Loranthaceae). PLoS ONE 2021, 16, 0256345. [Google Scholar]
- Vidal-Russell, R.; Nickrent, D.L. Evolutionary relationship in the showy mistletoe family (Loranthaceae). Am. J. Bot. 2008, 95, 1015–1029. [Google Scholar] [CrossRef]
- Watson, D.M. Mistletoe—A keystone resource in forests and woodlands worldwide. Annu. Rev. Ecol. Syst. 2001, 32, 219–249. [Google Scholar] [CrossRef]
- Engler, A. Loranthaceae. In Die Naturlichen Planzenfamilien, 3rd ed.; Engler, A., Prantl, K., Eds.; Wilhelm Engelmann: Leipzig, Germany, 1894; pp. 156–198. [Google Scholar]
- Tieghem, P. Sur le groupement des especes en genres dans les Loranthacees a calice gamosepale et antheres basifixes ou Dendrophthoees. Bull. Soc. Bot. Fr. 1895, 42, 241–272. [Google Scholar] [CrossRef][Green Version]
- Nickrent, D.L.; Malécot, V.; Vidal-Russell, R.; Der, J.P. A revised classification of Santalales. Taxon 2010, 59, 538–558. [Google Scholar] [CrossRef]
- Migahid, A.M. Flora of Saudi Arabia, 4th ed.; King Saud University Press: Riyadh, Saudi Arabia, 1996; Volume 2, pp. 68–69. [Google Scholar]
- Orfali, R. Pharmacological evaluation of secondary metabolites and their simultaneous determination in the arabian medicinal plant plicosepalus curviflorus using hptlc validated. J. Anal. Methods Chem. 2019, 2019, 7435909. [Google Scholar] [CrossRef]
- Nickrent, D.L.; Su, H.-J.; Lin, R.-Z.; Devkota, M.P.; Hu, J.-M.; Glatzel, G. Examining the needle in the haystack: Evolutionary relationships in the mistletoe genus Loranthus (Loranthaceae). Syst. Bot. 2021, 46, 403–415. [Google Scholar] [CrossRef]
- Plant of the World, Royal Botanical Gardens (Kew). 2022. Available online: https://powo.science.kew.org/ (accessed on 5 February 2022).
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Straub, S.C.K.; Parks, M.; Weitemier, K.; Fishbein, M.; Cronn, R.C.; Liston, A. Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. Am. J. Bot. 2012, 99, 349–364. [Google Scholar] [CrossRef]
- Mattick, J.S.; Gagen, M.J. The evolution of controlled multitasked gene networks: The role of introns and other noncoding RNAs in the development of complex organisms. Mol. Biol. Evol. 2001, 18, 1611–1630. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P. Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res. 2007, 35, e14. [Google Scholar] [CrossRef] [PubMed]
- Staden, R.; McLachlan, A.D. Codon preference and its use in identifying protein coding regions in long DNA sequences. Nucleic Acids Res. 1982, 10, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Li, B. Complete chloroplast genome sequence of Decaisnea insignis: Genome organization, genomic resources and comparative analysis. Sci. Rep. 2017, 7, 10073. [Google Scholar] [CrossRef]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, 253–259. [Google Scholar] [CrossRef]
- Bundschuh, R.; Altmuller, J.; Becker, C.; Nurnberg, P.; Gott, J.M. Complete characterization of the edited transcriptome of the mitochondrion of Physarum polycephalum using deep sequencing of RNA. Nucleic Acids Res. 2011, 39, 6044–6055. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef]
- Wicke, S.; Müller, K.F.; Claude, W.D.; Quandt, D.; Bellot, S.; Schneeweiss, G.M. Mechanistic model of evolutionary rate variation en route to a nonphotosynthetic lifestyle in plants. Proc. Natl. Acad. Sci. USA 2016, 113, 9045–9050. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Naumann, J. Molecular evolution of plastid genomes in parasitic flowering plants. Adv. Bot. Res. 2018, 85, 315–347. [Google Scholar]
- Bellot, S.; Cusimano, N.; Luo, S.; Sun, G.; Zarre, S.; Groger, A.; Temsch, E.; Renner, S.S. Assembled plastid and mitochondrial genomes, as well as nuclear genes, place the parasite family cynomoriaceae in the saxifragales. Genome Biol. Evol. 2016, 8, 2214–2230. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dorjee, T.; Chen, Y.; Gao, F.; Zhou, Y. The complete chloroplast genome sequencing analysis revealed an unusual IRs reduction in three species of subfamily Zygophylloideae. PLoS ONE 2022, 17, e0263253. [Google Scholar] [CrossRef] [PubMed]
- Schelkunov, M.I.; Nuraliev, M.S.; Logacheva, M.D. Rhopalocnemis phalloides has one of the most reduced and mutated plastid genomes known. PeerJ 2019, 7, e7500. [Google Scholar] [CrossRef]
- Su, H.-J.; Barkman, T.J.; Hao, W.; Jones, S.S.; Naumann, J.; Skippington, E.; Wafula, E.K.; Hu, J.-M.; Palmer, J.D.; dePamphilis, C.W. Novel genetic code and record-setting AT-richness in the highly reduced plastid genome of the holoparasitic plant Balanophora. Proc. Natl. Acad. Sci. USA 2018, 116, 934–943. [Google Scholar]
- Zong, D.; Zhang, Y.; Zou, X.; Li, D.; Duan, A.; He, C. Characterization of the complete chloroplast genomes of five Populus species from the western Sichuan plateau, southwest China: Comparative and phylogenetic analyses. PeerJ 2019, 7, e6386. [Google Scholar] [CrossRef]
- Nie, L.; Cui, Y.; Wu, L.; Zhou, J.; Xu, Z.; Li, Y.; Li, X.; Wang, Y.; Yao, H. Gene losses and variations in chloroplast genome of parasitic plant macrosolen and phylogenetic relationships within santalales. Int. J. Mol. Sci. 2019, 20, 5812. [Google Scholar] [CrossRef]
- Raman, G.; Park, S. The complete chloroplast genome sequence of Ampelopsis: Gene organization, comparative analysis, and phylogenetic relationships to other angiosperms. Front. Plant Sci. 2016, 7, 341. [Google Scholar] [CrossRef]
- Alzahrani, D.A.; Yaradua, S.S.; Albokhari, E.J.; Abba, A. Complete chloroplast genome sequences of Dipterygium glaucum and Cleome chrysantha and other Cleomaceae species, comparative analysis and phylogenetic relationships. Saudi J. Biol. Sci. 2021, 28, 2476–2490. [Google Scholar] [CrossRef]
- Wang, W.; Yu, H.; Wang, J.; Lei, W.; Gao, J.; Qiu, X.; Wang, J. The complete chloroplast genome sequences of the medicinal plant Forsythia suspensa (Oleaceae). Int. J. Mol. Sci. 2017, 18, 2288. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Park, H.; Lee, H.; Lee, B.; Lee, J. The complete plastome sequence of an Antarctic bryophyte Sanionia uncinata (Hedw.) Loeske. Int. J. Mol. Sci. 2018, 19, 709. [Google Scholar] [CrossRef] [PubMed]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Yang, A.-H.; Zhang, J.-J.; Yao, X.-H.; Huang, H.-W. Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplification in L. chinense. Am. J. Bot. 2011, 98, e123–e126. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Wang, S.; Zhou, S.-L. Polymorphic chloroplast microsatellite loci in Nelumbo (Nelumbonaceae). Am. J. Bot. 2012, 99, e240–e244. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Woeste, K.E.; Zhao, P. Completion of the chloroplast genomes of five Chinese juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 2017, 7, 1955. [Google Scholar] [CrossRef]
- Ruhsam, M.; Clark, A.; Finger, A.; Wulff, A.S.; Mill, R.R.; Thomas, P.I.; Gardner, M.F.; Gaudeul, M.; Ennos, R.A.; Hollingsworth, P.M. Hidden in plain view: Cryptic diversity in the emblematic Araucaria of New Caledonia. Am. J. Bot. 2016, 103, 888–898. [Google Scholar] [CrossRef]
- Sunil-Kumar, K.N.; Maruthi, K.R.; Alfarhan, A.H.; Rajakrishnan, R.; Thomas, J. Molecular fingerprinting of Helicanthus elastica (Desr.) Danser growing on five different hosts by RAPD. Saudi J. Biol. Sci. 2016, 23, 335–340. [Google Scholar] [CrossRef][Green Version]
- Friedrich, T.; Klaus, S.; Weiss, H. The proton–pumping respiratory complex I of bacteria and mitochondria and its homologue in chloroplasts. FEBS Lett. 1995, 367, 107–111. [Google Scholar] [CrossRef]
- Zhu, Z.X.; Wang, J.H.; Cai, Y.C.; Zhao, K.K.; Moore, M.J.; Wang, H.F. Complete plastome sequence of Erythropalum scandens (Erythropalaceae), an edible and medicinally important liana in China. Mitochondrial DNA Part B 2018, 3, 139–140. [Google Scholar] [CrossRef]
- Su, H.J.; Hu, J.M. The complete chloroplast genome of hemiparasitic flowering plant Schoepfia jasminodora. Mitochondrial DNA Part B 2016, 1, 767–769. [Google Scholar] [CrossRef]
- Frailey, D.C.; Chaluvadi, S.R.; Vaughn, J.N.; Goatney, C.G.; Bennetzen, J.L. Gene loss and genome rearrangement in the plastids of five Hemiparasites in the family Orobanchaceae. BMC Plant Biol. 2018, 18, 30. [Google Scholar] [CrossRef]
- Wicke, S.; Müller, K.F.; Claude, W.D.; Quandt, D.; Bellot, S.; Schneeweiss, G.M. Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the broomrape family. Plant Cell 2013, 25, 3711–3725. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.-L.; Su, T.-M.; Chaw, S.-M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 3650. [Google Scholar] [CrossRef]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W.; et al. Many Parallel Losses of infA from Chloroplast DNA during Angiosperm Evolution with Multiple Independent Transfers to the Nucleus. Plant Cell. 2001, 13, 645–658. [Google Scholar] [CrossRef]
- Goulding, S.E.; Wolfe, K.; Olmstead, R.; Morden, C. Ebb and flow of the chloroplast inverted repeat. Mol. Genet. Genom. 1996, 252, 195–206. [Google Scholar] [CrossRef]
- Sugiura, M. The chloroplast genome. In 10 Years Plant Molecular Biology; Springer: Dordrecht, The Netherlands, 1992; pp. 149–168. [Google Scholar]
- Daniell, H.; Lee, S.-B.; Grevich, J.; Saski, C.; Quesada-Vargas, T.; Guda, C.; Tomkins, J.; Jansen, R.K. Complete chloroplast genome sequences of Solanum bulbocastanum, Solanum lycopersicum and comparative analyses with other Solanaceae genomes. Theor. Appl. Genet. 2006, 112, 1503. [Google Scholar] [CrossRef]
- Yao, X.; Tan, Y.-H.; Liu, Y.-Y.; Song, Y.; Yang, J.-B.; Corlett, R.T. Chloroplast genome structure in Ilex (Aquifoliaceae). Sci. Rep. 2016, 6, 28559. [Google Scholar] [CrossRef]
- Wang, X.; Gussarova, G.; Ruhsam, M.; De Vere, N.; Metherell, C.; Hollingsworth, P.M.; Twyford, A.D. DNA barcoding a taxonomically complex hemiparasitic genus reveals deep divergence between ploidy levels but lack of species-level resolution. Ann. Bot. 2018, 10, ply026. [Google Scholar] [CrossRef]
- Alzahrani, D.A.; Yaradua, S.S.; Albokhari, E.J.; Abba, A. Complete chloroplast genome sequence of Barleria prionitis, comparative chloroplast genomics and phylogenetic relationships among Acanthoideae. BMC Genom. 2020, 21, 393. [Google Scholar] [CrossRef]
- Zhou, T.; Ruhsam, M.; Wang, J.; Zhu, H.; Li, W.; Zhang, X.; Xu, Y.; Xu, F.; Wang, X. The complete chloroplast genome of euphrasia regelii, pseudogenization of ndh genes and the phylogenetic relationships within orobanchaceae. Front. Genet. 2019, 10, 444. [Google Scholar] [CrossRef]
- Yaradua, S.S. Complete chloroplast genome sequence of justicia flava: Genome comparative analysis and phylogenetic relationships among acanthaceae. BioMed Res. Int. 2019, 17, 4370258. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Li, W.; Xie, X.; Lu, Y.; Liu, Y.; Jin, X.; Suo, Z. Phylogenetic resolution in Juglans based on complete chloroplast genomes and nuclear DNA sequences. Front. Plant Sci. 2017, 8, 1148. [Google Scholar] [CrossRef]
- Cabrera, J.F. Molecular Phylogeny and Historical Biogeography of Loranthaceae (Santalales) Inferred from Matk Sequence Data. Master’s Thesis, Southern Illinois University, Carbondale, IL, USA, 2002. [Google Scholar]
- Doyle, J.; Doyle, J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M.; Notes, A. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. Organellar genome DRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Wang, D. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Van Rossum, G.; Drake, F.L., Jr. Python Reference Manual; Centrum voor Wiskunde en Informatica: Amsterdam, The Netherlands, 1995. [Google Scholar]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | P. acaciae | P. curviflorus |
|---|---|---|
| Genome size (bp) | 120,181 | 121,086 |
| IRA (bp) | 22,323 | 22,476 |
| IRB (bp) | 22,323 | 22,476 |
| LSC (bp) | 69,497 | 69,947 |
| SSC (bp) | 6038 | 6187 |
| Total No of Genes | 106 | 108 |
| Total No of Unique Genes | 92 | 92 |
| rRNA | 4 | 4 |
| tRNA | 25 | 25 |
| Protein-Coding genes | 63 | 63 |
| A% | 31.50 | 31.34 |
| T (U) % | 31.93 | 31.89 |
| G% | 17.97 | 18.12 |
| C % | 18.59 | 18.65 |
| %GC | 36.6 | 36.8 |
| Gene | Strand | Location | Exon I (bp) | Intron I (bp) | Exon II (bp) | Intron II (bp) | Exon III (bp) |
|---|---|---|---|---|---|---|---|
| atpF | - | LSC | 145 | 801 | 401 | ||
| ycf3 | - | LSC | 124 | 762 | 230 | 768 | 153 |
| clpP | - | LSC | 71 | 767 | 294 | 675 | 229 |
| petB | + | LSC | 6 | 814 | 642 | ||
| rps12 | - | IRB | 56 | 515 | 229 | ||
| + | IRA | 229 | 515 | 56 | |||
| rpl2 | - | IRB | 391 | 654 | 431 | ||
| rpl16 | - | LSC | 303 | 486 | 9 | ||
| petD | + | LSC | 8 | 743 | 484 | ||
| trnL-UAA | + | LSC | 35 | 299 | 55 | ||
| trnA-UGC | - | IRA | 55 | 342 | 39 | ||
| + | IRB | 39 | 342 | 55 |
| Gene | Strand | Location | Exon I (bp) | Intron I (bp) | Exon II (bp) | Intron II (bp) | Exon III (bp) |
|---|---|---|---|---|---|---|---|
| atpF | - | LSC | 145 | 792 | 410 | ||
| rpoC1 | - | LSC | 432 | 830 | 1617 | ||
| ycf3 | - | LSC | 124 | 743 | 230 | 772 | 153 |
| clpP | - | LSC | 71 | 752 | 294 | 524 | 229 |
| rpl2 | - | IRB | 391 | 654 | 440 | ||
| rps12 | - | IRB | 56 | 507 | 229 | ||
| + | IRA | 229 | 507 | 56 | |||
| rpl16 | - | LSC | 9 | 893 | 81 | ||
| petB | + | LSC | 6 | 864 | 642 | ||
| trnL-UAA | + | LSC | 35 | 290 | 55 | ||
| trnA-UGC | - | IRA | 55 | 350 | 37 | ||
| + | IRB | 37 | 350 | 55 |
| SSR Type | Repeat Unit | Plicosepalus acacia | Plicosepalus curviflorus | Scurrula chingii | Taxillus chinensis | Loranthus europaeus | Dendrophthoe pentandra | Nuytsia floribunda | Elytranthe albida |
|---|---|---|---|---|---|---|---|---|---|
| Mono | A | 67 | 62 | 59 | 49 | 44 | 50 | 47 | 60 |
| C | 2 | 2 | 4 | 7 | 6 | 6 | 3 | 6 | |
| G | 1 | 0 | 2 | 2 | 2 | 0 | 3 | 2 | |
| T | 69 | 70 | 87 | 86 | 67 | 78 | 69 | 87 | |
| Di | AG/CT | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 |
| AT/AT | 10 | 6 | 13 | 6 | 7 | 5 | 12 | 3 | |
| Tri | AAG/CTT | 2 | 2 | 2 | 2 | 2 | 2 | 0 | 0 |
| AAT/ATT | 4 | 1 | 1 | 1 | 1 | 3 | 2 | 1 | |
| ATC/ATG | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | |
| Tetra | AAAG/CTTT | 3 | 3 | 1 | 1 | 2 | 2 | 0 | 3 |
| AAAT/ATTT | 4 | 2 | 1 | 0 | 4 | 5 | 3 | 1 | |
| ACAG/CTGT | 1 | 0 | 1 | 1 | 1 | 1 | 0 | 1 | |
| AGGG/CCCT | 0 | 2 | 2 | 0 | 2 | 2 | 0 | 0 | |
| AAGT/ACTT | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | |
| AATT/AATT | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | |
| AATC/ATTG | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | |
| AAAC/GTTT | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | |
| AACC/GGTT | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | |
| AGAT/ATCT | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | |
| AATG/ATTC | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | |
| Penta | AAAAT/ATTTT | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| AATAT/ATATT | 0 | 3 | 0 | 1 | 2 | 0 | 0 | 0 | |
| Total | 164 | 155 | 175 | 157 | 143 | 155 | 142 | 169 |
| Order | Family | Accession Number | Organism |
|---|---|---|---|
| Santalales | Loranthaceae | NC_053563 | Scurrula chingii (W.C.Cheng) H.S.Kiu |
| MN080717 | Taxillus chinensis (DC.) Danser | ||
| MN080718.1 | Helixanthera parasitica Lour. | ||
| MT987630 | Loranthus europaeus Jacq. | ||
| MT987635.1 | Loranthus pseudo-odoratus Lingelsh. | ||
| NC_039376 | Macrosolen cochinchinensis (Lour.) Tiegh. | ||
| NC_045107 | Dendrophthoe pentandra (L.) Miq. | ||
| NC_045108 | Elytranthe albida (Blume) Blume | ||
| NC_058837 | Taxillus pseudochinensis (Yamam.) Danser | ||
| NC_058840 | Taxillus tsaii S.T.Chiu | ||
| NC_058859 | Cecarria obtusifolia (Merr.) Barlow | ||
| NC_058862.1 | Loranthus guizhouensis H.S.Kiu | ||
| NC_058868 | Moquiniella rubra (A.Spreng.) Balle | ||
| NC_058869 | Nuytsia floribunda (Labill.) R.Br. ex G.Don | ||
| NC_058858.1 | Loranthus delavayi Tiegh. | ||
| Santalaceae | NC_027960 | Osyris alba L. | |
| Schoepfiaceae | NC_034228 | Schoepfia jasminodora Siebold & Zucc. | |
| Viscaceae | KT003925.1 | Viscum album L. | |
| NC_027829 | Viscum minimum Harv. | ||
| Erythropalaceae | NC_036759.1 | Erythropalum scandens Blume | |
| Lamiales | Orobanchaceae | NC_001568.1 | Epifagus virginiana (L.) Barton |
| Solanales | Solanaceae | NC_001879 | Nicotiana tabacum L. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AL-Juhani, W.; Al Thagafi, N.T.; Al-Qthanin, R.N. Gene Losses and Plastome Degradation in the Hemiparasitic Species Plicosepalus acaciae and Plicosepalus curviflorus: Comparative Analyses and Phylogenetic Relationships among Santalales Members. Plants 2022, 11, 1869. https://doi.org/10.3390/plants11141869
AL-Juhani W, Al Thagafi NT, Al-Qthanin RN. Gene Losses and Plastome Degradation in the Hemiparasitic Species Plicosepalus acaciae and Plicosepalus curviflorus: Comparative Analyses and Phylogenetic Relationships among Santalales Members. Plants. 2022; 11(14):1869. https://doi.org/10.3390/plants11141869
Chicago/Turabian StyleAL-Juhani, Widad, Noha T. Al Thagafi, and Rahmah N. Al-Qthanin. 2022. "Gene Losses and Plastome Degradation in the Hemiparasitic Species Plicosepalus acaciae and Plicosepalus curviflorus: Comparative Analyses and Phylogenetic Relationships among Santalales Members" Plants 11, no. 14: 1869. https://doi.org/10.3390/plants11141869
APA StyleAL-Juhani, W., Al Thagafi, N. T., & Al-Qthanin, R. N. (2022). Gene Losses and Plastome Degradation in the Hemiparasitic Species Plicosepalus acaciae and Plicosepalus curviflorus: Comparative Analyses and Phylogenetic Relationships among Santalales Members. Plants, 11(14), 1869. https://doi.org/10.3390/plants11141869