
Gene Co-Expression Network Tools and Databases for Crop Improvement

 , , , and
, , , and
Abstract
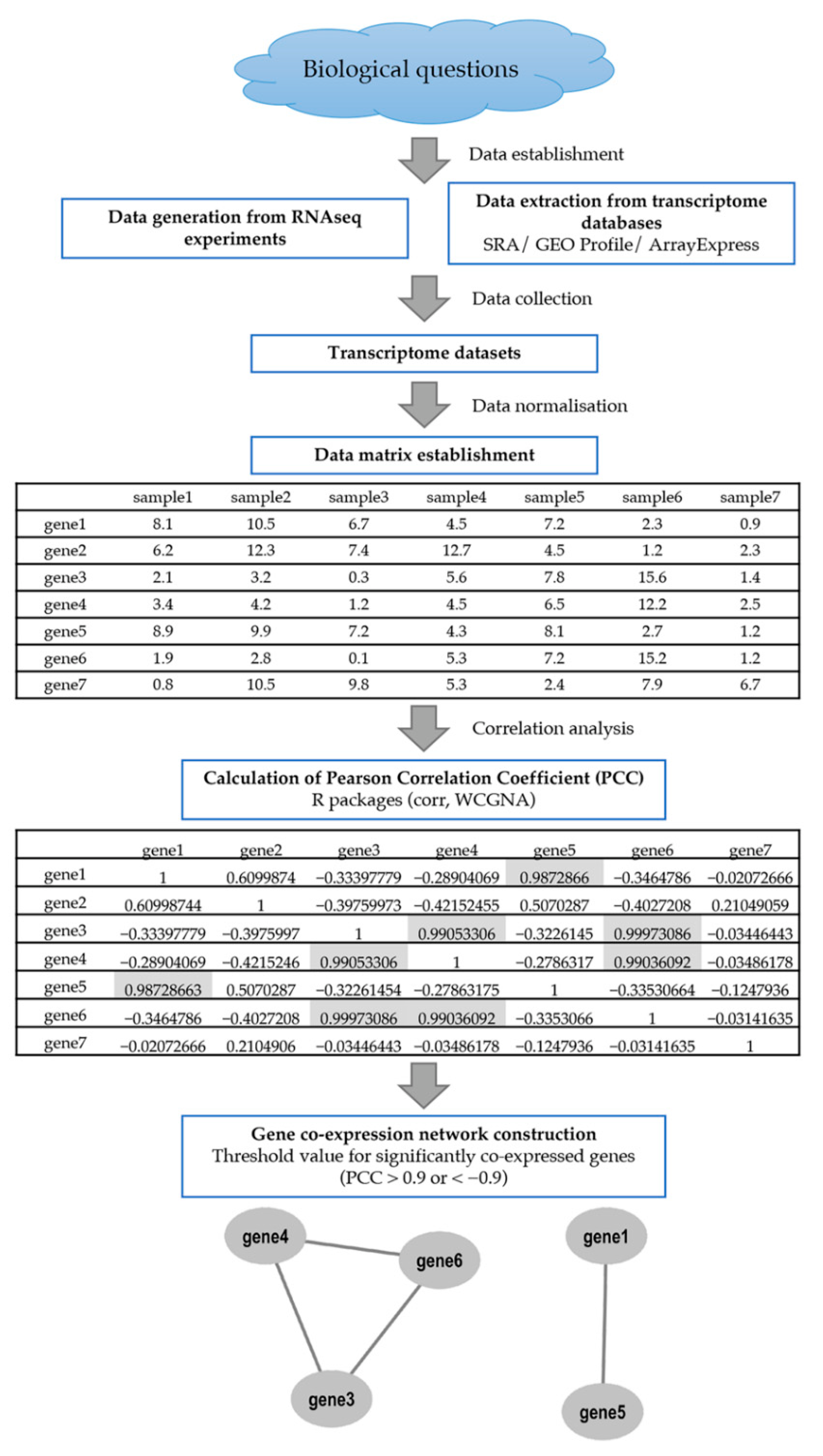
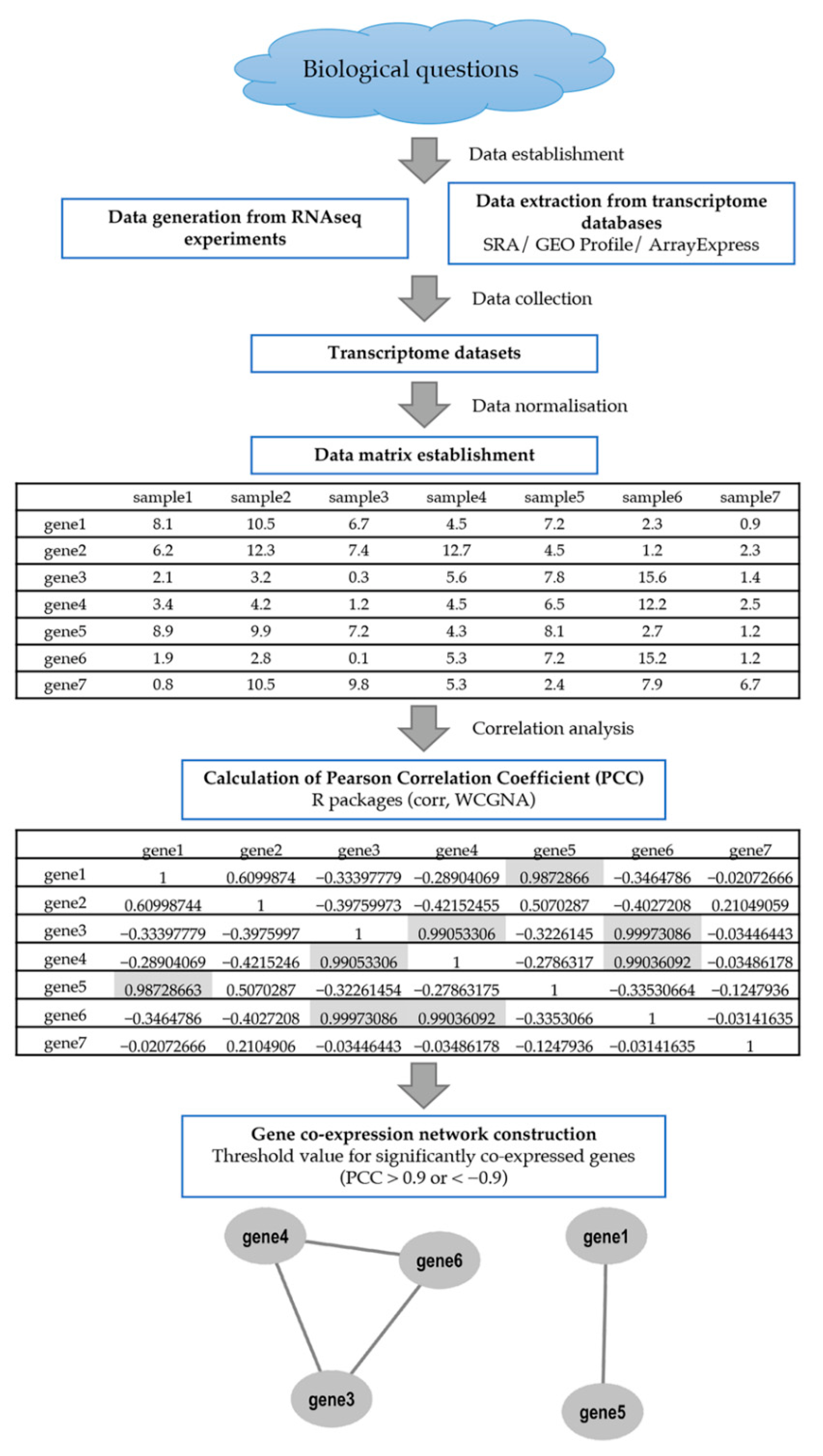
:1. Introduction
2. Overview on Co-Expression Tools for Analysing the Gene Co-Expression Network in Crops
2.1. Web-Based Tool to Construct Gene Co-Expression Network
2.1.1. CORNET
2.1.2. CoExpNetViz and PlaNet
2.1.3. RECoN
2.1.4. webCEMiTool
2.2. Command-Line Tools
2.2.1. Weighted Correlation Network Analysis (WGCNA)
2.2.2. Petal
2.2.3. LSTrAP
2.2.4. COGENT
2.2.5. GWENA
2.2.6. Juxtapose
3. Overview of Current Gene Co-Expression Network Databases in Plants
3.1. Oryza sativa
3.2. Zea mays
3.3. Sorghum bicolor
3.4. Vitis vinifera
3.5. Solanum lycopersicum
3.6. Malus domestica
3.7. Phyllostachys edulis
3.8. Camelia sinensis
3.9. Brassica napus
3.10. Multiple Species Gene Co-Expression Network Databases
3.10.1. ATTED-II
3.10.2. PLANEX
3.10.3. PlantNexus
3.10.4. Co-Expression Network Toolkit (CoNekT)
3.10.5. CoCoCoNet
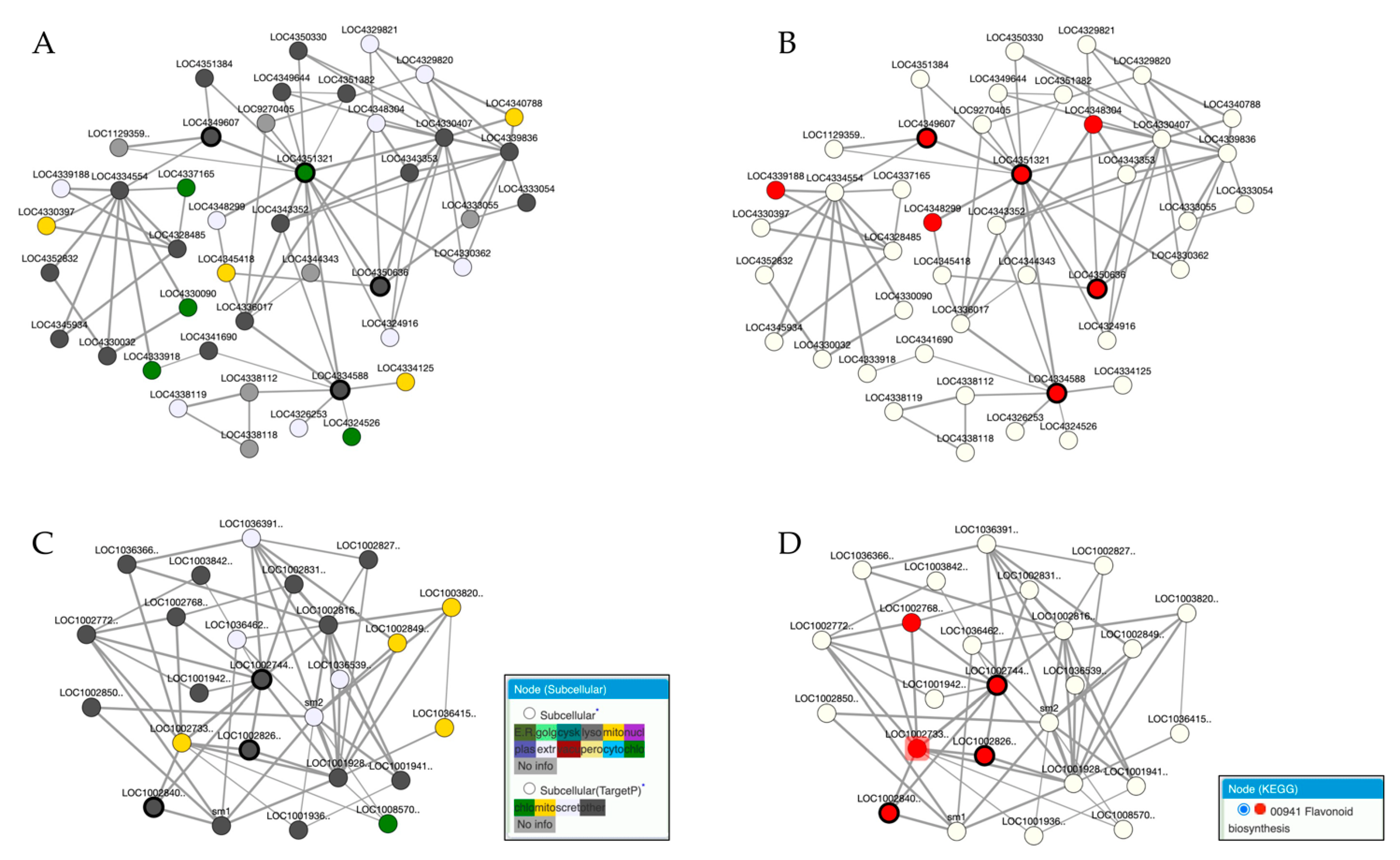
4. Case Study: Application of Co-Expression Networks in Biological Pathway Identification
5. Perspective, Challenges and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, W.; Lu, Z.; Xiong, Y.; Yao, J. Genome-Wide Identification and Co-Expression Network Analysis of the OsNF-Y Gene Family in Rice. Crop J. 2017, 5, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Xie, T.; Chen, C.; Luan, A.; Long, J.; Li, C.; Ding, Y.; He, Y. Genome-Wide Organization and Expression Profiling of the R2R3-MYB Transcription Factor Family in Pineapple (Ananas Comosus). BMC Genom. 2017, 18, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahan, R.; Zawora, C.; Wight, H.; Sittmann, J.; Wang, W.; Mount, S.M.; Liu, Z. Consensus Coexpression Network Analysis Identifies Key Regulators of Flower and Fruit Development in Wild Strawberry. PLANT Physiol. 2018, 178, 202–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets-Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athar, A.; Fullgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress Update—from Bulk to Single-Cell Expression Data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Vendramin, S.; Shi, L.; McGinnis, K.M. Construction and Optimization of a Large Gene Coexpression Network in Maize Using RNA-Seq Data. Plant Physiol. 2017, 175, 568–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Lu, L.; Jiao, B.; Liang, C. Systematic Discovery of Novel and Valuable Plant Gene Modules by Large-Scale RNA-Seq Samples. Bioinformatics 2019, 35, 361–364. [Google Scholar] [CrossRef]
- Sircar, S.; Parekh, N. Meta-Analysis of Drought-Tolerant Genotypes in Oryza Sativa: A Network-Based Approach. PLoS ONE 2019, 14, e0216068. [Google Scholar] [CrossRef] [Green Version]
- Aoki, K.; Ogata, Y.; Shibata, D. Approaches for Extracting Practical Information from Gene Co-Expression Networks in Plant Biology. Plant Cell Physiol. 2007, 48, 381–390. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Maslov, S. Pan- and Core- Network Analysis of Co-Expression Genes in a Model Plant. Sci. Rep. 2016, 6, 38956. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Lund, J.; Kiraly, M.; Duke, K.; Jiang, M.; Stuart, J.M.; Eizinger, A.; Wylie, B.N.; Davidson, G.S. A Gene Expression Map for Caenorhabditis Elegans. Science 2001, 293, 2087–2092. [Google Scholar] [CrossRef] [Green Version]
- Parsana, P.; Ruberman, C.; Jaffe, A.E.; Schatz, M.C.; Battle, A.; Leek, J.T. Addressing Confounding Artifacts in Reconstruction of Gene Co-Expression Networks. GENOME Biol. 2019, 20, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Usadel, B.; Obayashi, T.; Mutwil, M.; Giorgi, F.M.; Bassel, G.W.; Tanimoto, M.; Chow, A.; Steinhauser, D.; Persson, S.; Provart, N.J. Co-Expression Tools for Plant Biology: Opportunities for Hypothesis Generation and Caveats. Plant Cell Environ. 2009, 32, 1633–1651. [Google Scholar] [CrossRef]
- Emamjomeh, A.; Saboori Robat, E.; Zahiri, J.; Solouki, M.; Khosravi, P. Gene Co-Expression Network Reconstruction: A Review on Computational Methods for Inferring Functional Information from Plant-Based Expression Data. Plant Biotechnol. Rep. 2017, 11, 71–86. [Google Scholar] [CrossRef]
- Ruprecht, C.; Persson, S. Co-Expression of Cell Wall-Related Genes: New Tools and Insights. Front. Plant Sci. 2012, 3, 83. [Google Scholar] [CrossRef] [Green Version]
- Beiki, H.; Nejati-Javaremi, A.; Pakdel, A.; Masoudi-Nejad, A.; Hu, Z.L.; Reecy, J.M. Large-Scale Gene Co-Expression Network as a Source of Functional Annotation for Cattle Genes. BMC Genom. 2016, 17, 846. [Google Scholar] [CrossRef] [Green Version]
- Yong, Y.B.; Li, W.Q.; Wang, J.M.; Zhang, Y.; Lu, Y.M. Identification of Gene Co-Expression Networks Involved in Cold Resistance of Lilium Lancifolium. Biol. Plant. 2018, 62, 287–298. [Google Scholar] [CrossRef]
- Ashari, K.-S.; Abdullah-Zawawi, M.-R.; Harun, S.; Mohamed-Hussein, Z.-A. Reconstruction of the Transcriptional Regulatory Network in Arabidopsis Thaliana Aliphatic Glucosinolate Biosynthetic Pathway. Sains Malays. 2018, 47, 2993–3002. [Google Scholar] [CrossRef]
- Da, L.; Liu, Y.; Yang, J.; Tian, T.; She, J.; Ma, X.; Xu, W.; Su, Z. AppleMDO: A Multi-Dimensional Omics Database for Apple Co-Expression Networks and Chromatin States. Front. Plant Sci. 2019, 10, 1333. [Google Scholar] [CrossRef]
- Harun, S.; Afiqah-Aleng, N.; Karim, M.B.; Ul Amin, M.A.; Kanaya, S.; Mohamed-Hussein, Z.-A. Potential Arabidopsis Thaliana Glucosinolate Genes Identified from the Co-Expression Modules Using Graph Clustering Approach. PeerJ 2021, 9, e11876. [Google Scholar] [CrossRef]
- Harun, S.; Rohani, E.R.; Ohme-Takagi, M.; Goh, H.-H.; Mohamed-Hussein, Z.-A. ADAP Is a Possible Negative Regulator of Glucosinolate Biosynthesis in Arabidopsis Thaliana Based on Clustering and Gene Expression Analyses. J. Plant Res. 2021, 134, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Harun, S.; Afiqah-Aleng, N.; Abdul Hadi, F.I.; Lam, S.D.; Mohamed-Hussein, Z.-A. Identification of Potential Genes Encoding Protein Transporters in Arabidopsis Thaliana Glucosinolate (GSL) Metabolism. Life 2022, 12, 326. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.C.J. Network Aggregation Improves Gene Function Prediction of Grapevine Gene Co-Expression Networks. PLANT Mol. Biol. 2020, 103, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Tzfadia, O.; Diels, T.; De Meyer, S.; Vandepoele, K.; Aharoni, A.; van De Peer, Y. CoExpNetViz: Comparative Co-Expression Networks Construction and Visualization Tool. Front. Plant Sci. 2016, 6, 1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proost, S.; Krawczyk, A.; Mutwil, M. LSTrAP: Efficiently Combining RNA Sequencing Data into Co-Expression Networks. BMC Bioinform. 2017, 18, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardozo, L.E.; Russo, P.S.T.; Gomes-Correia, B.; Araujo-Pereira, M.; Sepúlveda-Hermosilla, G.; Maracaja-Coutinho, V.; Nakaya, H.I. WebCEMiTool: Co-Expression Modular Analysis Made Easy. Front. Genet. 2019, 10, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Ruiz, S.; Gil-Martínez, A.L.; Cisterna, A.; Jurado-Ruiz, F.; Reynolds, R.H.; Cookson, M.R.; Hardy, J.; Ryten, M.; Botía, J.A. CoExp: A Web Tool for the Exploitation of Co-Expression Networks. Front. Genet. 2021, 12, 630187. [Google Scholar] [CrossRef]
- Itkin, M.; Heinig, U.; Tzfadia, O.; Bhide, A.J.; Shinde, B.; Cardenas, P.D.; Bocobza, S.E.; Unger, T.; Malitsky, S.; Finkers, R.; et al. Biosynthesis of Antinutritional Alkaloids in Solanaceous Crops Is Mediated by Clustered Genes. Science 2013, 341, 175–179. [Google Scholar] [CrossRef]
- de Bodt, S.; Carvajal, D.; Hollunder, J.; van den Cruyce, J.; Movahedi, S.; Inzé, D. CORNET: A User-Friendly Tool for Data Mining and Integration. Plant Physiol. 2010, 152, 1167–1179. [Google Scholar] [CrossRef] [Green Version]
- Proost, S.; Mutwil, M. Planet: Comparative Co-Expression Network Analyses for Plants. Methods Mol. Biol. 2017, 1533, 213–227. [Google Scholar] [CrossRef]
- Krishnan, A.; Gupta, C.; Ambavaram, M.M.R.; Pereira, A. RECoN: Rice Environment Coexpression Network for Systems Level Analysis of Abiotic-Stress Response. Front. Plant Sci. 2017, 8, 1640. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Petereit, J.; Smith, S.; Harris, F.C.; Schlauch, K.A. Petal: Co-Expression Network Modelling in R. BMC Syst. Biol. 2016, 10, 181–258. [Google Scholar] [CrossRef] [Green Version]
- Bozhilova, L.V.; Pardo-Diaz, J.; Reinert, G.; Deane, C.M. COGENT: Evaluating the Consistency of Gene Co-Expression Networks. Bioinformatics 2021, 37, 1928–1929. [Google Scholar] [CrossRef]
- Lemoine, G.G.; Scott-Boyer, M.P.; Ambroise, B.; Périn, O.; Droit, A. GWENA: Gene Co-Expression Networks Analysis and Extended Modules Characterization in a Single Bioconductor Package. BMC Bioinform. 2021, 22, 267. [Google Scholar] [CrossRef]
- Ovens, K.; Maleki, F.; Eames, B.F.; McQuillan, I. Juxtapose: A Gene-Embedding Approach for Comparing Co-Expression Networks. BMC Bioinform. 2021, 22, 125. [Google Scholar] [CrossRef]
- De Bodt, S.; Hollunder, J.; Nelissen, H.; Meulemeester, N.; Inze, D. Methods Interactions, Regulatory Interactions, Gene Associations and Functional Annotations. New Phytol. 2012, 195, 707–720. [Google Scholar] [CrossRef]
- Mutwil, M.; Klie, S.; Tohge, T.; Giorgi, F.M.; Wilkins, O.; Campbell, M.M.; Fernie, A.R.; Usadel, B.; Nikoloski, Z.; Persson, S. PlaNet: Combined Sequence and Expression Comparisons across Plant Networks Derived from Seven Species. Plant Cell 2011, 23, 895–910. [Google Scholar] [CrossRef] [Green Version]
- Saelens, W.; Cannoodt, R.; Saeys, Y. A Comprehensive Evaluation of Module Detection Methods for Gene Expression Data. Nat. Commun. 2018, 9, 1090. [Google Scholar] [CrossRef]
- Du, J.; Wang, S.; He, C.; Zhou, B.; Ruan, Y.L.; Shou, H. Identifcation of Regulatory Networks and Hub Genes Controlling Soybean Seed Set and Size Using RNA Sequencing Analysis. J. Exp. Bot. 2017, 68, 1955–1972. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Lv, L.; Meng, C.; Zhou, C.; Fu, J.; Shen, X.; Zhang, C.; Li, Y. Genome-Wide Analysis of Abscisic Acid Biosynthesis, Catabolism, and Signaling in Sorghum Bicolor under Saline-Alkali Stress. Biomolecules 2019, 9, 823. [Google Scholar] [CrossRef] [Green Version]
- Esposito, S.; Aversano, R.; Bradeen, J.; D’Amelia, V.; Villano, C.; Carputo, D. Coexpression Gene Network Analysis of Cold-Tolerant Solanum Commersonii Reveals New Insights in Response to Low Temperatures. Crop Sci. 2021, 61, 3538–3550. [Google Scholar] [CrossRef]
- Jia, X.; Feng, H.; Bu, Y.; Ji, N.; Lyu, Y.; Zhao, S. Comparative Transcriptome and Weighted Gene Co-Expression Network Analysis Identify Key Transcription Factors of Rosa Chinensis ‘Old Blush’ after Exposure to a Gradual Drought Stress Followed by Recovery. Front. Genet. 2021, 12, 690264. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, M.; Chen, J.; Qing, C.; He, S.; Zou, C.; Yuan, G.; Yang, C.; Peng, H.; Pan, G.; et al. GWAS and WGCNA Uncover Hub Genes Controlling Salt Tolerance in Maize (Zea mays L.) Seedlings. Theor. Appl. Genet. 2021, 134, 3305–3318. [Google Scholar] [CrossRef]
- Xia, L.; Zou, D.; Sang, J.; Xu, X.; Yin, H.; Li, M.; Wu, S.; Hu, S.; Hao, L.; Zhang, Z. Rice Expression Database (RED): An Integrated RNA-Seq-Derived Gene Expression Database for Rice. J. Genet. Genom. 2017, 44, 235–241. [Google Scholar] [CrossRef]
- Sato, Y.; Namiki, N.; Takehisa, H.; Kamatsuki, K.; Minami, H.; Ikawa, H.; Ohyanagi, H.; Sugimoto, K.; Itoh, J.-I.; Antonio, B.A.; et al. RiceFREND: A Platform for Retrieving Coexpressed Gene Networks in Rice. Nucleic Acids Res. 2013, 41, D1214–D1221. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; You, Q.; Yan, H.; Xu, W.; Su, Z. MCENet: A Database for Maize Conditional Co-Expression Network and Network Characterization Collaborated with Multi-Dimensional Omics Levels. J. Genet. Genom. 2018, 45, 351–360. [Google Scholar] [CrossRef]
- Tian, T.; You, Q.; Zhang, L.; Yi, X.; Yan, H.; Xu, W.; Su, Z. SorghumFDB: Sorghum Functional Genomics Database with Multidimensional Network Analysis. Database 2016, baw099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, D.C.J.; Sweetman, C.; Drew, D.P.; Ford, C.M. VTCdb: A Gene Co-Expression Database for the Crop Species Vitis Vinifera (Grapevine). BMC Genom. 2013, 14, 882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narise, T.; Sakurai, N.; Obayashi, T.; Ohta, H.; Shibata, D. Co-Expressed Pathways DataBase for Tomato: A Database to Predict Pathways Relevant to a Query Gene. BMC Genom. 2017, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhao, H.; Xu, W.; You, Q.; Yan, H.; Gao, Z.; Su, Z. Co-Expression Gene Network Analysis and Functional Module Identification in Bamboo Growth and Development. Front. Genet. 2018, 9, 574. [Google Scholar] [CrossRef]
- Zhang, R.; Ma, Y.; Hu, X.; Chen, Y.; He, X.; Wang, P.; Chen, Q.; Ho, C.-T.; Wan, X.; Zhang, Y.; et al. TeaCoN: A Database of Gene Co-Expression Network for Tea Plant (Camellia Sinensis). BMC Genom. 2020, 21, 461. [Google Scholar] [CrossRef]
- Chao, H.; Li, T.; Luo, C.; Huang, H.; Ruan, Y.; Li, X.; Niu, Y.; Fan, Y.; Sun, W.; Zhang, K.; et al. Brassicaedb: A Gene Expression Database for Brassica Crops. Int. J. Mol. Sci. 2020, 21, 5831. [Google Scholar] [CrossRef]
- Yim, W.C.; Yu, Y.; Song, K.; Jang, C.S.; Lee, B.M. PLANEX: The Plant Co-Expression Database. BMC Plant Biol. 2013, 13, 83. [Google Scholar] [CrossRef] [Green Version]
- Obayashi, T.; Aoki, Y.; Tadaka, S.; Kagaya, Y.; Kinoshita, K. ATTED-II in 2018: A Plant Coexpression Database Based on Investigation of the Statistical Property of the Mutual Rank Index. Plant Cell Physiol. 2017, 59, e3. [Google Scholar] [CrossRef]
- Zhou, Y.; Sukul, A.; Mishler-Elmore, J.W.; Faik, A.; Held, M.A. PlantNexus: A Gene Co-Expression Network Database and Visualization Tool for Barley and Sorghum. Plant Cell Physiol. 2022, 63, 565–572. [Google Scholar] [CrossRef]
- Proost, S.; Mutwil, M. CoNekT: An Open-Source Framework for Comparative Genomic and Transcriptomic Network Analyses. Nucleic Acids Res. 2018, 46, W133–W140. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Shah, M.; Ballouz, S.; Crow, M.; Gillis, J. CoCoCoNet: Conserved and Comparative Co-Expression across a Diverse Set of Species. Nucleic Acids Res. 2021, 48, W566–W571. [Google Scholar] [CrossRef]
- Ballouz, S.; Weber, M.; Pavlidis, P.; Gillis, J. EGAD: Ultra-Fast Functional Analysis of Gene Networks. Bioinformatics 2017, 33, 612–614. [Google Scholar] [CrossRef]
- Park, H.L.; Yoo, Y.; Bhoo, S.H.; Lee, T.H.; Lee, S.W.; Cho, M.H. Two Chalcone Synthase Isozymes Participate Redundantly in Uv-Induced Sakuranetin Synthesis in Rice. Int. J. Mol. Sci. 2020, 21, 3777. [Google Scholar] [CrossRef]
- Park, S.-I.; Park, H.-L.; Bhoo, S.-H.; Lee, S.-W.; Cho, M.-H. Biochemical and Molecular Characterization of the Rice Chalcone Isomerase Family. Plants 2021, 10, 2064. [Google Scholar] [CrossRef]
- Ovens, K.; Eames, B.F.; McQuillan, I. Comparative Analyses of Gene Co-Expression Networks: Implementations and Applications in the Study of Evolution. Front. Genet. 2021, 12, 695399. [Google Scholar] [CrossRef]
- Fukushima, A.; Nishizawa, T.; Hayakumo, M.; Hikosaka, S.; Saito, K.; Goto, E.; Kusano, M. Exploring Tomato Gene Functions Based on Coexpression Modules Using Graph Clustering and Differential. Genome Anal. 2012, 158, 1487–1502. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, S.; Ogata, Y.; Suda, K.; Kurabayashi, A.; Suzuki, T.; Yamamoto, N.; Iijima, Y.; Tsugane, T.; Fujii, T.; Konishi, C.; et al. Coexpression Analysis of Tomato Genes and Experimental Verification of Coordinated Expression of Genes Found in a Functionally Enriched Coexpression Module. DNA Res. 2010, 17, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Baldoni, E.; Frugis, G.; Martinelli, F.; Benny, J.; Paffetti, D.; Buti, M. A Comparative Transcriptomic Meta-Analysis Revealed Conserved Key Genes and Regulatory Networks Involved in Drought Tolerance in Cereal Crops. Int. J. Mol. Sci. 2021, 22, 13062. [Google Scholar] [CrossRef]
- Reshef, D.N.; Reshef, Y.A.; Finucane, H.K.; Grossman, S.R.; McVean, G.; Turnbaugh, P.J.; Lander, E.S.; Mitzenmacher, M.; Sabeti, P.C. Detecting Novel Associations in Large Data Sets. Science 2011, 334, 1518–1524. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Wang, X. Application of the Gini Correlation Coefficient to Infer Regulatory Relationships in Transcriptome Analysis. Plant Physiol. 2012, 160, 192–203. [Google Scholar] [CrossRef] [Green Version]
- Lim, P.K.; Zheng, X.; Goh, J.C.; Mutwil, M. Exploiting Plant Transcriptomic Databases: Resources, Tools, and Approaches. Plant Commun. 2022, 3, 100323. [Google Scholar] [CrossRef]
- Kontio, J.A.J.; Rinta-Aho, M.J.; Sillanpää, M.J. Estimating Linear and Nonlinear Gene Coexpression Networks by Semiparametric Neighborhood Selection. Genetics 2020, 215, 597–607. [Google Scholar] [CrossRef]
- Rao, X.; Dixon, R.A. Co-Expression Networks for Plant Biology: Why and How. Acta Biochim. Biophys. Sin. 2019, 51, 981–988. [Google Scholar] [CrossRef]
- Wang, Y.X.R.; Waterman, M.S.; Huang, H. Gene Coexpression Measures in Large Heterogeneous Samples Using Count Statistics. Proc. Natl. Acad. Sci. USA 2014, 111, 16371–16376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.X.R.; Liu, K.; Theusch, E.; Rotter, J.I.; Medina, M.W.; Waterman, M.S.; Huang, H. Generalized Correlation Measure Using Count Statistics for Gene Expression Data with Ordered Samples. Bioinformatics 2018, 34, 617–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Li, P.; Zhu, M.; Wang, X.; Lu, J.; Yu, T. Nonlinear Network Reconstruction from Gene Expression Data Using Marginal Dependencies Measured by DCOL. PLoS ONE 2016, 11, e0158247. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Langfelder, P.; Horvath, S. Comparison of Co-Expression Measures: Mutual Information, Correlation, and Model Based Indices. BMC Bioinform. 2012, 13, 328. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Zhang, Y.; Hu, W.; Tan, H.; Wang, X. Inferring Nonlinear Gene Regulatory Networks from Gene Expression Data Based on Distance Correlation. PLoS ONE 2014, 9, e0087446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Ye, X.; Feng, W.; Zhang, Q.; Han, Y.; Liu, Y.; Li, Y.; Wei, Y. Distance Correlation Application to Gene Co-Expression Network Analysis. BMC Bioinform. 2022, 23, 81. [Google Scholar] [CrossRef] [PubMed]
- Albert, E.; Gricourt, J.; Bertin, N.; Bonnefoi, J.; Pateyron, S.; Tamby, J.-P.; Bitton, F.; Causse, M. Genotype by Watering Regime Interaction in Cultivated Tomato: Lessons from Linkage Mapping and Gene Expression. Theor. Appl. Genet. 2016, 129, 395–418. [Google Scholar] [CrossRef]
- Guo, T.; Yang, J.; Li, D.; Sun, K.; Luo, L.; Xiao, W.; Wang, J.; Liu, Y.; Wang, S.; Wang, H.; et al. Integrating GWAS, QTL, Mapping and RNA-Seq to Identify Candidate Genes for Seed Vigor in Rice (Oryza sativa L.). Mol. Breed. 2019, 39, 87. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Types | Co-Expression Network Tool | Descriptions | References |
|---|---|---|---|
| Web-based tool | CORNET 2.0 https://bioinformatics.psb.ugent.be/cornet (accessed on 11 March 2022) | An integrating tool for plant co-expression network | [29] |
| http://bioinformatics.psb.ugent.be/webtools/coexpr/ (accessed on 11 March 2022) | A comparative co-expression network construction and visualisation | [24] | |
| PlaNet www.gene2function.de (accessed on 11 March 2022) | A tool for comparative co-expression network analyses | [30] | |
| RECoN https://plantstress-pereira.uark.edu/RECoN/ (accessed on 11 March 2022) | A co-expression tool to identify co-expressed genes in abiotic stress response | [31] | |
| webCemiTool https://cemitool.sysbio.tools/ (accessed on 11 March 2022) | A web-based tool to identify co-expression modules in a given co-expression network | [26] | |
| CoExp https://rytenlab.com/coexp (accessed on 11 March 2022) | A web tool for the exploitation of co-expression networks | [27] | |
| Command-line based tool & require installation | WGCNA https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/ (accessed on 9 April 2022) | An R package for performing weighted correlation network analysis | [32] |
| petal https://github.com/julipetal/petalNet (accessed on 9 April 2022) | An R package for co-expression network modelling | [33] | |
| LSTrAP https://github.molgen.mpg.de/proost/LSTrAP (accessed on 9 April 2022) | A pipeline to construct co-expression networks from RNA-seq data | [25] | |
| COGENT https://github.com/lbozhilova/COGENT (accessed on 9 March 2022) | An R package to construct a gene co-expression network without the need for annotation or external validation data. | [34] | |
| GWENA https://bioconductor.org/packages/release/bioc/html/GWENA.html (accessed on 9 March 2022) | An R package developed to extend the analysis of gene co-expression network | [35] | |
| Juxtapose https://github.com/klovens/juxtapose (accessed on 9 March 2022) | A tool to compare gene co-expression networks (GCNs) | [36] |
| Plant Species | Databases | Descriptions | Statistical Methods | References |
|---|---|---|---|---|
| Oryza sativa | Rice Expression database http://expression.ic4r.org/co-search (accessed on 29 March 2022) | A repository of gene expression profiles and co-expression network. | PCC | [45] |
| RiceFrend https://ricefrend.dna.affrc.go.jp/ (accessed on 10 March 2022) | A gene co-expression database in rice based on an extensive collection of microarray data derived from various tissues/organs at different stages of growth and development under natural field conditions. | PCC & Mutual Rank | [46] | |
| Zea mays | MCENet http://bioinformatics.cau.edu.cn/MCENet/ (accessed on 10 March 2022) | A database for maize co-expression networks. | PCC and Mutual Rank | [47] |
| Maize gene co-expression network database https://www.bio.fsu.edu/mcginnislab/mcn/main_page.php (accessed on 10 March 2022) | A gene co-expression network database for maize. | PCC, KCC, SCC and Mutual Information | [6] | |
| Sorghum bicolor | Sorghum Functional Genomics Database (SorghumFDB) http://structuralbiology.cau.edu.cn/sorghum/index.html (accessed on 15 March 2022) | A sorghum database to predict gene function. | PCC and Mutual Rank | [48] |
| Vitis vinifera | VTCdb: ViTis Co-expression DataBase http://vtcdb.adelaide.edu.au/Home.aspx (accessed on 15 March 2022) | A database for co-expressed genes in grapes. | PCC, SCC, Highest Reciprocal and Mutual Rank | [49] |
| Solanum lycopersicum | Co-expressed pathways database for tomato http://cox-path-db.kazusa.or.jp/tomato/ (accessed on 10 March 2022) | A database for co-expressed genes in tomatoes. | PCC, ORA (p-value), GSEA (p-value, percentile-scores) | [50] |
| Phyllostachys edulis | BambooNET http://bioinformatics.cau.edu.cn/bamboo/ (accessed on 10 March 2022) | A database of co-expression networks with functional modules for bamboo. | PCC and Mutual Rank | [51] |
| Malus domestica | AppleMDO http://bioinformatics.cau.edu.cn/AppleMDO/ (accessed on 10 March 2022) | A multi-dimensional omics database for apple co-expression networks and chromatin states. | PCC and Mutual Rank | [19] |
| Camellia sinesis | TeaCoN http://teacon.wchoda.com/ (accessed on 10 March 2022) | A database of gene co-expression network for tea plants. | PCC | [52] |
| Brassica napus | BrassicaEDB https://brassica.biodb.org/ (accessed on 13 March 2022) | A database of gene co-expression network and expression profiles for Brassica crops. | PCC and weight value | [53] |
| Multiple crop species | PLANEX http://planex.plantgenomicslab.org/ (accessed on 13 March 2022) | A plant gene co-expression database obtained from GEO NCBI. | PCC, Gene enrichment analysis (Cohen’s Kappa) | [54] |
| ATTED-II https://atted.jp/ (accessed on 13 March 2022) | A plant co-expression database. | PCC, SCC and Mutual Rank | [55] | |
| PlantNexus http://planex.plantgenomicslab.org/ (accessed on 13 March 2022) | A gene co-expression network database for barley and sorghum. | [56] | ||
| CoNekT-P https://conekt.sbs.ntu.edu.sg/ (accessed on 19 May 2022) | An online platform that allows users to browse co-expression networks and perform comparative GCN analysis across different crop species (rice, maize, tomato) and others plant species. | HRR and HCCA | [57] | |
| CoCoCoNet https://milton.cshl.edu/CoCoCoNet/ (accessed on 19 May 2022) | A comparative gene co-expression network portal for a diverse range of species including plants, humans and animals. | SCC | [58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zainal-Abidin, R.-A.; Harun, S.; Vengatharajuloo, V.; Tamizi, A.-A.; Samsulrizal, N.H. Gene Co-Expression Network Tools and Databases for Crop Improvement. Plants 2022, 11, 1625. https://doi.org/10.3390/plants11131625
Zainal-Abidin R-A, Harun S, Vengatharajuloo V, Tamizi A-A, Samsulrizal NH. Gene Co-Expression Network Tools and Databases for Crop Improvement. Plants. 2022; 11(13):1625. https://doi.org/10.3390/plants11131625
Chicago/Turabian StyleZainal-Abidin, Rabiatul-Adawiah, Sarahani Harun, Vinothienii Vengatharajuloo, Amin-Asyraf Tamizi, and Nurul Hidayah Samsulrizal. 2022. "Gene Co-Expression Network Tools and Databases for Crop Improvement" Plants 11, no. 13: 1625. https://doi.org/10.3390/plants11131625
APA StyleZainal-Abidin, R.-A., Harun, S., Vengatharajuloo, V., Tamizi, A.-A., & Samsulrizal, N. H. (2022). Gene Co-Expression Network Tools and Databases for Crop Improvement. Plants, 11(13), 1625. https://doi.org/10.3390/plants11131625