1. Introduction
The genus
Piper belongs to the Piperaceae family, considered as one of the largest genera among angiosperms and one of the most abundant species in tropical forests [
1]. Several species have high economic value, such as
Piper nigrum (black pepper), the representative species of the Piperaceae family used worldwide as a condiment having medicinal properties.
Piper species have been widely investigated, and a number of physiological activity components have been isolated, which can be characterized into the typical classes of alkaloids, amides, chalcones, dihydrochalcones, flavanones, lignans, neolignans, propenylphenols, steroids, and terpenes [
1]. However, only a few chemical investigations of
P. crocatum have been reported, which were limited to sterols, lignans, and neolignans [
2,
3,
4]. In China, people have used the dried stem of
Piper kadsura (Choisy) Ohwi as a traditional Chinese medicine to treat asthma and rheumatism for hundreds of years. The species of
P. crocatum is commonly used in folk medicine in Indonesia to treat various diseases [
2]. Many species of the Piperaceae family have been extensively studied for their potential antitumor, antimicrobial, and antifungal properties [
2,
5]. Therefore, the chemical constitution of
P. crocatum still needs to be investigated because it is a source of novel natural products with potential activities. The metabolic characteristics of neoplastic and non-neoplastic cells are considerably different. However, non-neoplastic cells predominantly depend on ATP/energy produced by pyruvate oxidation in the mitochondria, and each molecular glucose oxidized generates 36 ATPs; whereas proliferating cancer cells predominantly rely on aerobic glycolysis in the cytoplasm, and up to four ATPs are produced for each glucose molecule [
6]. Pyruvate dehydrogenase complex and pyruvate dehydrogenase kinase (PDK) are key mitochondrial enzymes in the metabolic pathway of glucose, and their interaction regulates the proportion between aerobic respiration and the Warburg effect [
7,
8]. Because all tumors rely on metabolic alterations for growth, metastasis, and survival, atypical pathways may be potential targets of antineoplastic drugs.
3. Discussion
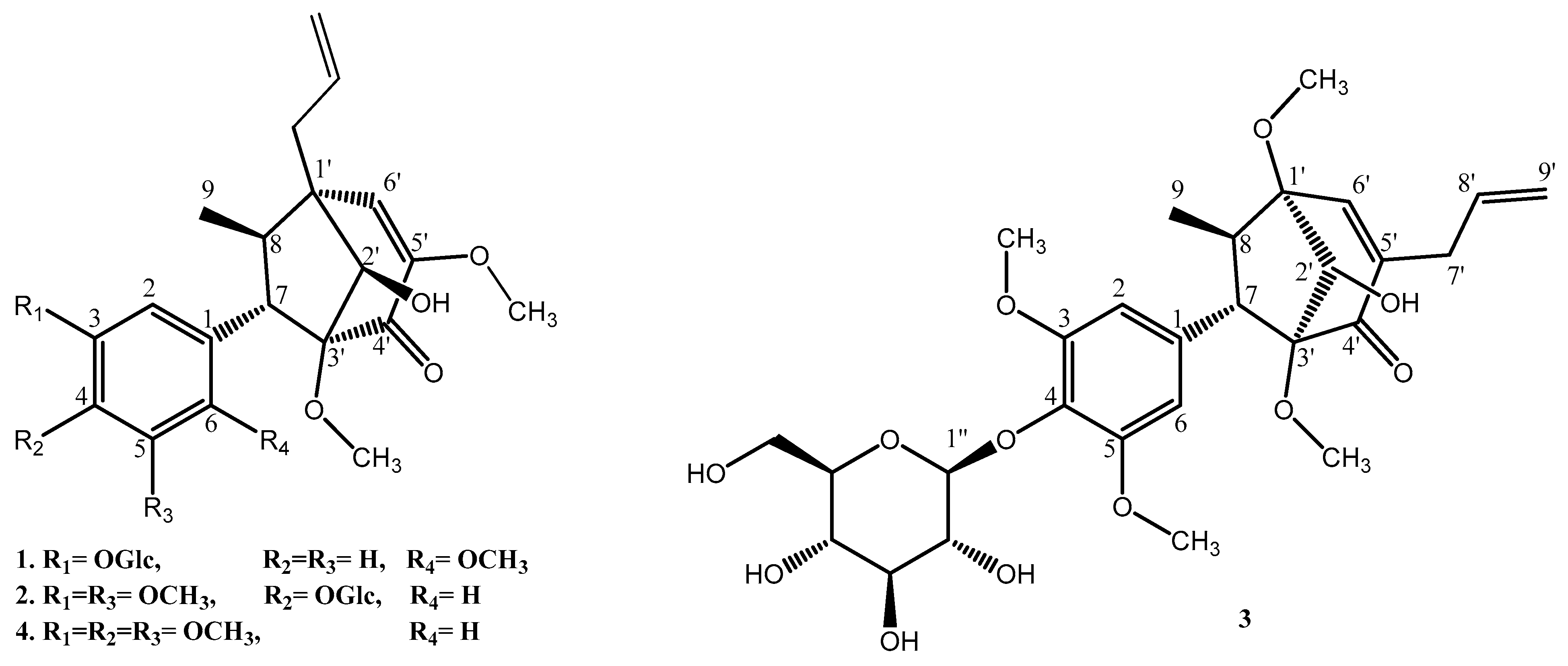
Compound
1 was isolated as a white amorphous powder. Its molecular formula was established as C
27H
36O
11 using HR-ESI-MS. The
1H NMR spectrum (
Table 1) showed the presence of an ABX system of aromatic protons at δ
H 6.47 (t, J = 2.0 Hz, H-2), 6.48 (dd, J = 8.0, 2.0 Hz, H-4), and 6.87 (d, J = 8.0 Hz, H-5), as well as an allyl group suggested from the COSY spectrum at δ
H 2.30 (dd, J = 13.7, 7.0 Hz, H-7′a), 2.63 (dd, J = 13.7, 7.0 Hz, H-7′b), 5.14 (td, J = 10.0 Hz, H-9′a), 5.27 (d, J = 17.0 Hz, H-9′b), and 5.93 (td, J = 17.0, 7.0 Hz, H-8′), and 3.24 (m, H-7)/2.13 (p, J = 7.0 Hz, H-8), and a signal assigned to the anomeric proton of the glucosyl moiety at δ
H 4.84 (d, J = 7.2 Hz, H-1″). Other proton signals suggested three methoxyl groups at δ
H 3.16 (s, 3′-OMe), 3.56 (s, 5′-OMe), and 3.64 (s, 6-OMe), as well as a methyl group attached to the aliphatic carbon at δ
H 1.15 (d, J = 7.0 Hz, H-9). The
13C NMR spectrum indicated the presence of six aromatic carbons at δ
C 112.4 (C-2), 114.3 (C-5), 121.4 (C-4), 132.4 (C-1), 145.2 (C-3), and 148.0 (C-6) and a glucosyl moiety at δ
C 60.4 (C-6″), 69.5 (C-4″), 73.1 (C-2″), 76.8 (C-3″), 76.9 (C-5″), and 99.6 (C-1″). Other signals, such as a carbinol carbon at δ
C 78.1 (C-2′); an allyl group at δ
C 34.1 (C-7′), 118.0 (C-9′), and 135.3 (C-8′); olefinic carbons at δ
C 126.6 (C-6′) and 151.6 (C-5′); two quaternary carbons at δ
C 48.6 (C-1′) and 96.1 (C-3′); and a carbonyl group at δ
C 192.4 (C-4′), indicated a bicyclo[3.2.1]octane derivative, which was similar to compound
4. Therefore, both
1H and
13C NMR spectra demonstrated that compound
1 was a bicyclo[3.2.1]octane lignan derivative. The connection between aromatic ring, glycosyl group, and bicyclo[3.2.1]octane of compound
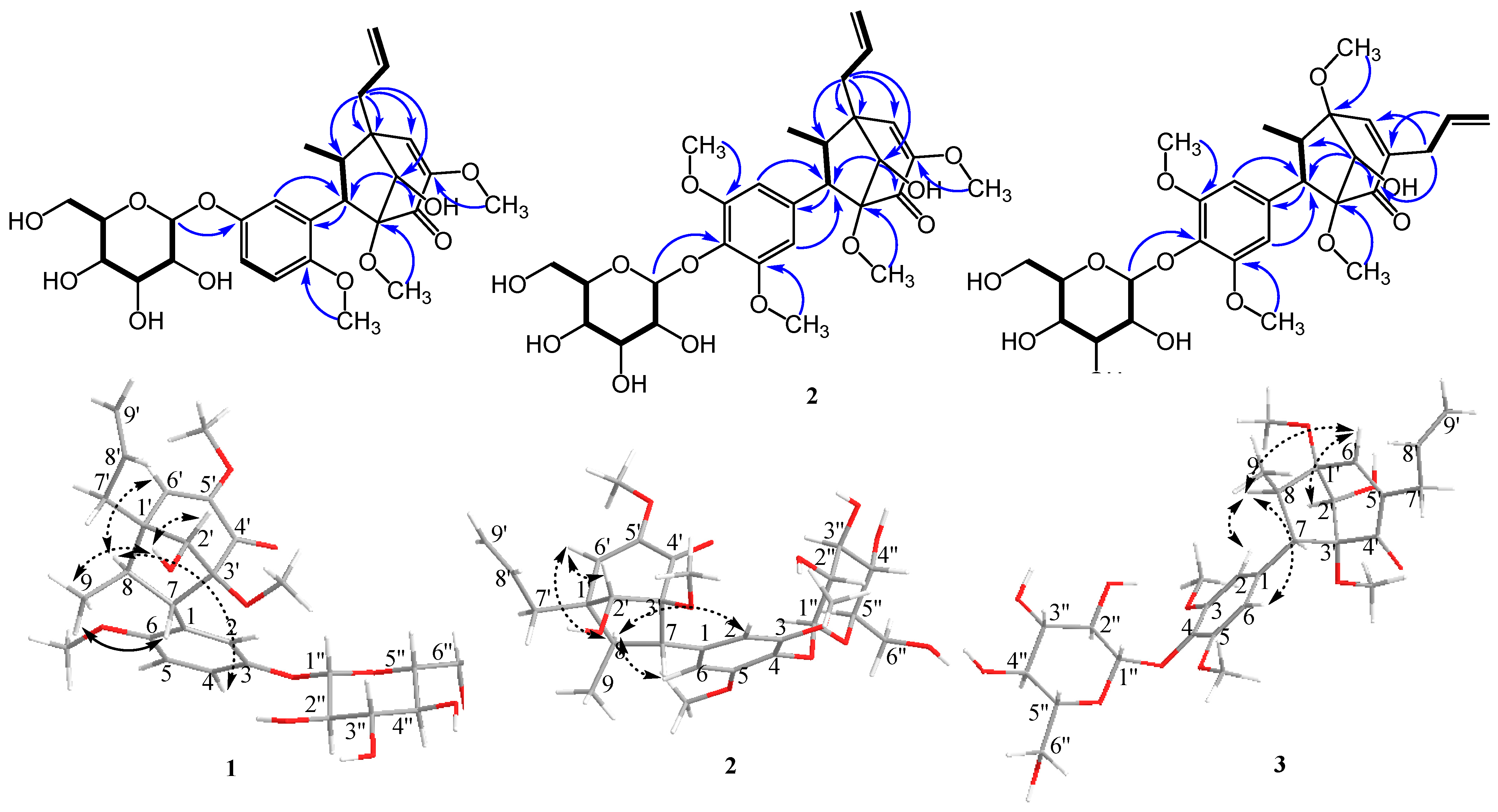
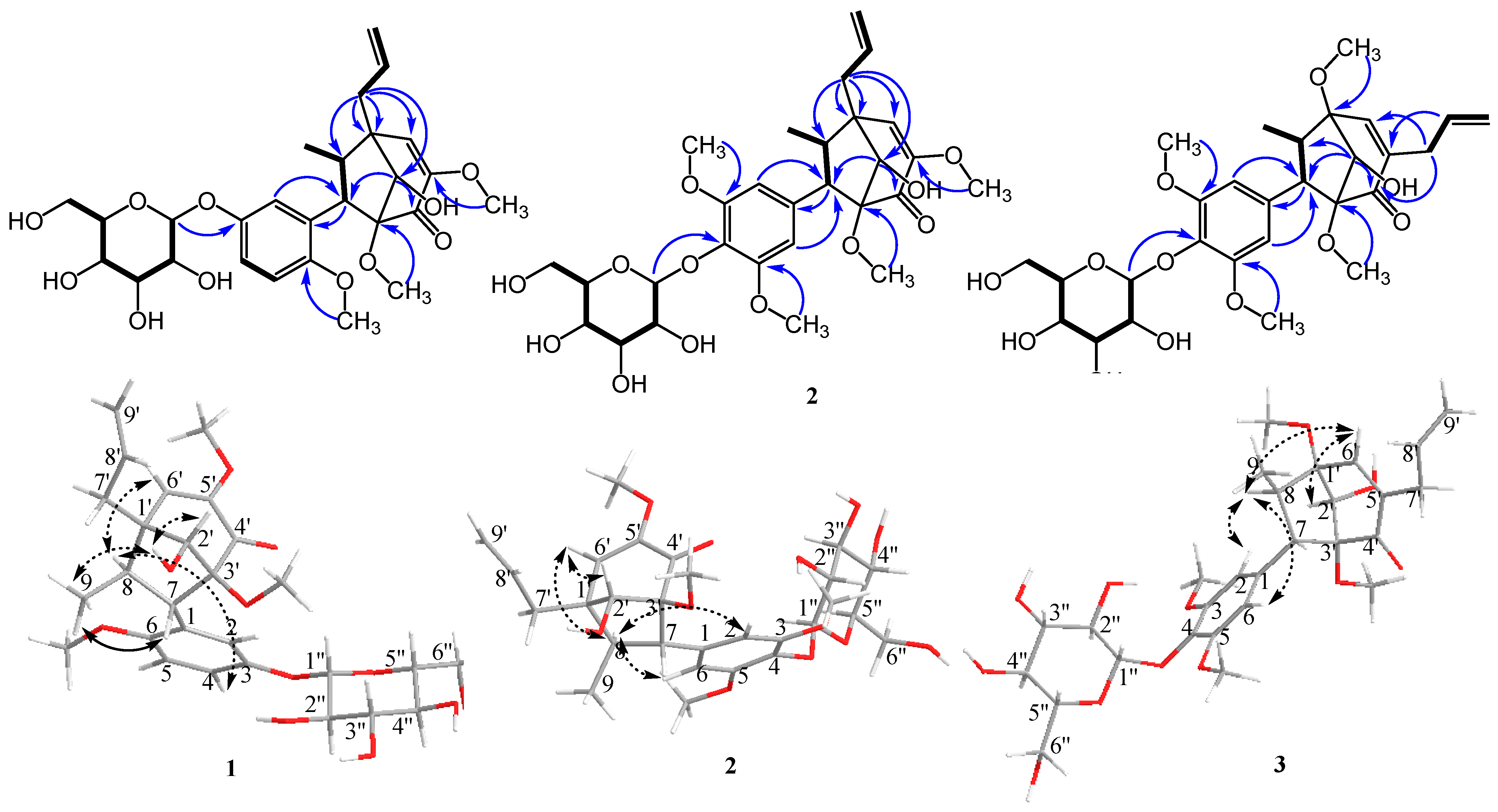
1 was unambiguously assigned based on HMBC spectra (
Figure 2). Key HMBC correlations between H-2′ (δ
H 3.81)/C-4′ (δ
C 192.4) and C-7 (δ
C 57.9) indicate the connection between the C
3-bicyclooctane moiety at C-3′. Similarly, H-7′ (δ
H 2.30 and 2.63)/C-1′ (δ
C 48.6), C-2′ (δ
C 78.1), C-6′ (δ
C 126.6), and C-8 (δ
C 47.8) further supported the presence of the C
3-bicyclooctane moiety at C-1′ (δ
C 48.6) and the assigned position of an allyl group at C-1′. The correlation between H-1″ (δ
H 4.84) and 145.2 (C-3) indicated that the glycosyl group was attached at C-7 of the aromatic ring. These data led to the identification of a guianin-type 7.1′,8.3′-connected neolignan skeleton of compound
1.
The relative stereochemistry of methyl (H-9) and aryl groups was revealed from the key ROESY correlation between H-9 and H-7, and no correlation was observed between H-9 and aromatic ortho-protons (H-2 and H-6). However, a correlation between H-8 and H-4 was observed. Therefore, the trans-relationship of methyl (H-9) and aryl groups was confirmed [
4]. Additionally, the correlations between H-2′, –OH-2′, and H-9 were observed, which indicated the endo conformation of the hydroxy group on C-2′. The correlation between H-9 and –OH-2′, which indicated the relative configuration of the hydroxy group, suggested endo configuration with the methyl (H-9) group. The relative configuration of
1 was further determined based on the reported chemical shifts of H-7, H-6′, and methyl at C-8. In 1′S, 2′S, 3′R configuration series, H-7 was determined to be resonated at δ
H 2.6, H-6′ at δ
H 5.7, and methyl between δ
H 0.8 and 0.9, whereas in 1′R, 2′R, 3′S configuration series, the resonances for H-7, H-6′, and methyl protons appeared at δ
H 3.3, 6.2, and 1.25–1.28, respectively [
9]. For our study, the chemical shifts of H-7, H-6′, and H-8 methyl were δ
H 3.24, 6.29, and 1.15, respectively. The absolute configuration of compound
1 was deduced to be (1′,2′R,3′S,7S,8R)-neolignan, and two adjacent torsion angles were measured (C2′–C3′–C4′–C5′ = −37.7° and C2′–C1′–C6′–C5′ = 36.5°) and compared with those reported in the literature [
10] (see
Supplementary Materials). Based on the abovementioned data, the structure of compound
1 was determined as (1′R,2′R,3′S,7S,8R)-∆
5′
,8′-2′-hydroxy-6,3′,5′-trimethoxyl-4′-oxo-8.1′7.3′-neolignan-3-
O-β-
d-glucopyranoside, and the compound was named pipcroside A.
Compound
2 was isolated as a colorless gum. The molecular formula was established as C
28H
38O
12 using HR-ESI-MS. The
1H and
13C spectroscopic data (
Table 1) of compound
2 were determined to be identical to those of compound
4 except for that of the glucose moiety, and the signal was assigned to the anomeric proton of the glucosyl group at δ
H 4.66 (d, J = 7.2 Hz, H-1″). Thus, the absolute configuration of the C
3-bicyclooctane moiety of compound
2 was the same as that of compound
4, and the absolute configurations were confirmed using X-ray [
3]. Based on the abovementioned data, the absolute configuration of compound
2 was determined as (1′R,2′R,3′S,7S,8R)-∆
5′
,8′-2′-hydroxy-3,5,3′,5′-tetramethoxyl-4′-oxo-8.1′7.3′-neolignan-4-
O-β-
d-glucopyranoside, and the compound was named pipcroside B.
Compound
3 was isolated as a colorless gum. The molecular formula was established as C
28H
38O
12 using HR-ESI-MS. The
1H NMR spectrum data (
Table 1) indicated the existence of four methoxyl groups, including two symmetric methoxyl groups substituted on the aromatic ring at δ
H 3.79 (s, 3-OMe and 5-OMe), two methoxyl groups attached to aliphatic carbons at δ
H 3.34 (s, 1′-OMe) and 3.49 (s, 3′-OMe), and an allyl group at δ
H 5.86 (dd, J = 17.0, 10.0 Hz, H-8′), 5.11 (ddd, J = 17.0, 3.0, 2.0 Hz, H-9′), 2.92 (ddd, J = 16.0, 7.0, 1.0 Hz, H-7′), and 2.99 (ddd, J = 16.0, 7.0, 1.0 Hz, H-7′). The
1H NMR chemical shift of the C-8 methyl group at δ
H 1.28 (d, J = 7.0 Hz, H-9) and other compounds indicated a trans-relationship between the C-7 aryl and C-8 methyl groups. However, the
13C NMR spectrum showed one quaternary carbon δ
C 86.2 (C-1′) along with olefinic carbons at δ
C 139.5 (C-5′) and 156.5 (C-6′), which were considerably different from the other three compounds. The HMBC correlations further confirmed these connections among the bicyclooctane moiety through colo1′-OMe (δ
H 3.34)/C-1′ (δ
C 86.2), which indicated the deshielding effect of C-1′ (δ
C 86.2) compared with the other three compounds. Together with H-7′ (δ
H 2.92 and 2.99), C-5′ (δ
C 139.5), C-6′ (δ
C 156.5), and C-4′ (δ
C 199.1) confirmed that the allyl group was connected to an olefinic carbon C-5′ (δ
C 139.5). Subsequently, by comparing with the reported literature [
6,
11], compound
3 was considered to comprise a macrophyllin-type bicyclo[3.2.1]octanoid neolignan glucoside compound, and two adjacent torsion angles were measured (C2′–C3′–C4′–C5′ = 39.3° and C2′–C1′–C6′–C5′ = −34.8°) and compared with the reported literature (Coy-Barrera et al. 2012) (see
Supplementary Materials). Hence, the relative configuration for the C
3-bicyclooctane moiety of compound
3 was determined to be (1′R,2′S,3′R,7S,8R)-∆
5′
8′-2′-hydroxy-3,5,1′,3′-tetramethoxyl-4′-oxo-8.1′7.3′-neolignan-4-
O-β-
d-glucopyranoside, and the compound was named pipcroside C.
Reports from the literature were compared [
12,
13,
14] for the total synthesis of bicyclo[3.2.1]octanoid neolignans. The speculated biosynthetic pathways are in the
Supplementary Materials.
PDH Inhibition Effect
PDH is essential in the pathway of glucose metabolism and ATP production. Suppression of PDH reduces the aerobic oxidation of pyruvate and promotes its transformation to lactate in cytoplasm. Additionally, decreased flux of pyruvate via PDH diminishes the amount of acetyl-CoA entering into the TCA cycle. Accordingly, defects in both PDH and oxidative phosphorylation activities result in lactic acidosis and initiation of the Warburg effect [
7]. The activity of PDH is mainly controlled by reversible phosphorylation of the E1α subunit on residue sites serine 293 (Site 1), 300 (Site 2), and 232 (Site 3), which are mediated by PDK and pyruvate dehydrogenase phosphatase (PDP) phosphorylation of E1α by PDKs, which inactivates PDH, whereas PDP exhibits active/reactive catalytic activity by dephosphorylating the complex [
15]. Compounds
2 and
3 were evaluated for their inhibitory effect against PDHE1α S300 phosphorylation; we discuss the phosphorylation inhibitory effect of the E1α subunit on residue site serine 300 (Site 2). Fluorescent image results suggested that compounds
2 and
3 inhibited phosphorylation of E1α S300 at 60 µM with better effect than that of dichloroacetate (DCA) at 5 mM concentration; compound
2 showed the strongest effect among all three compounds (
Figure 3).
The IC
50 values of compounds
2 and
3 were rather high; that is, 99.82 and 80.25 µM, respectively. For the positive control DCA, the IC
50 values were 1.03 µM with 0.1%, 0.97 µM with 0.6%, and 1.06 µM with 1.0% concentration, respectively (
Figure 4).
Although previous research on DCA was promising, its application to anticancer treatment is limited by its non-specificity, low potency, and requirement for high doses to exhibit therapeutic effects, which always leads to peripheral neurological toxicity. The position exchange of the allyl group on C-1′ and methoxyl group on C-5′ resulted in a higher IC50 value difference between compounds 2 and 3 and revealed a better inhibitory effect of compound 2 than that of compound 3 and DCA in fluorescent imaging. Although compounds 2 and 3 possessed a rather high toxicity, the consumption of these two compounds was 83 times lower than that of DCA. The described type of neolignan compounds is one of the major components of Piper plants.
4. Materials and Methods
4.1. General Experimental Procedures
Optical rotations were determined using a Jasco DIP-370 automatic polarimeter (Jasco, Tokyo, Japan). The NMR spectra were recorded using a BRUKER AVANCE III 600 (1H, 600 MHz; 13C, 150 MHz) (Bruker Biospin GmbH, Karlsruhe, Germany), with tetramethylsilane (TMS) as an internal standard. Heteronuclear multiple quantum correlation (HMQC), heteronuclear multiple bond correlation (HMBC), rotating frame nuclear overhauser effect spectroscopy (ROESY), and 1H–1H correlation spectroscopy (COSY) spectra were recorded using a pulsed field gradient. The HR-ESI-MS spectra were obtained by using an Aglient 1200 LC-MSD Trap spectrometer (Agilent, Santa Clara, CA, USA). Preparative HPLC was performed using a GILSON 321 pump, 151 UV–VIS detector (Gilson, VILLIERS-LE-BEL, France), and RStech HECTOR-M C18 column (5-micron, 250 × 21.2 mm) (RS Tech Crop, Chungju, South Korea). Column chromatography was performed using a silica gel (Kieselgel 60, 70–230, and 230–400 mesh, Merck, Darmstadt, Germany), YMC C-18 resins, and thin layer chromatography (TLC) was performed using pre-coated silica-gel 60 F254 and RP-18 F254S plates (both 0.25 mm, Merck, Darmstadt, Germany). The spots were detected under UV light and using 10% H2SO4. The adjacent torsion angles were measured by Chemdraw 3D (version 17.1).
4.2. Plant Material
Dried leaves of P. crocatum were collected from Cilendek Timur, Bogor, West Java, Indonesia, in August 2016 and taxonomically identified (Identification number: 1714/IPH.1.01/If.07/VIII/2016) by the staff at the herbarium Laboratory, Research Center for Biology, Indonesian Institute of Sciences, Cibinong, West Java, Indonesia. A voucher specimen (BBRC-SMPLS-003) was deposited at the Herbarium of Aretha Medika Utama, Biomolecular and Biomedical Research Center, Bandung, West Java, Indonesia.
4.3. Extraction and Isolation
The dried leaves of P. crocatum (2.6 kg) were reflux extracted with MeOH (5 L × 3 times). The total extraction (400.0 g) of MeOH was suspended in deionized water and partitioned with hexane, and water fraction. Then the water fraction was partitioned sequentially with EtOAc and BuOH, yielding EtOAc (1A, 16.1 g), BuOH (1B, 65.0 g). The EtOAc fraction was subjected to a silica gel column chromatography with a gradient of CHCl3-MeOH-H2O (10:1:0, 9:1:0, 8:1:0, 6:1:0.1, 5:1:0.1, 4:1:0.1, 3:1:0.1, 2:1:0.1, MeOH 2.0 L for each step) to give 11 fractions (Fr. 1A-1-1A-11). Fractions 5 and 6 were combined and isolated with a gradient of MeOH–water (1:2, 1:1, and MeOH) by MPLC using a YMC C18 column to give 8 fractions (Fr. 2A-1-2A-8). Subfraction 2A-8 was isolated by prep-HPLC (MeOH: H2O = 60:40 retention time = 41.6 min) to give compound 4 (100.0 mg). The BuOH fraction was subjected to silica-gel column chromatography with a gradient of CHCl3-MeOH-H2O (10:1:0, 9:1:0, 8:1:0, 6:1:0.1, 5:1:0.1, 4:1:0.1, 3:1:0.1, 2:1:0.1, MeOH 5.0 L for each step) to give 11 fractions (Fr. 2B-1-2B-11). Fractions 2B-2 and 2B-3 were combined and isolated with a gradient of MeOH–H2O (1:2, 1:1, and MeOH) by MPLC using a YMC C18 column to give 12 fractions (Fr. 1C-1-1C-12). The fraction 1C-10 was separated by a Sephadex LH-20 column and eluted by MeOH and its subfraction was isolated by prep-HPLC (MeOH: H2O = 45:55 retention time = 55.1 min and then ACN: H2O = 20:80 retention time = 62.3 min) to give compound 3 (4.0 mg). The fraction 1C-12 was separated by a Sephadex LH-20 column and eluted by MeOH and its subfraction was isolated by prep-HPLC (MeOH: H2O = 80:20 retention time = 95.2 min) to give compound 1 (5.0 mg). Fractions 2B-4 and 2B-5 were combined and isolated with a gradient of MeOH–H2O (1:2, 1:1, and MeOH) by MPLC using a YMC C18 column to give 13 fractions (1D-1-1D-13). The fraction 1D-13 was separated by a Sephadex LH-20 column and eluted by MeOH, and its subfraction was isolated by prep-HPLC (MeOH: H2O = 50:50 retention time = 44.5 min) to give compound 2 (60.0 mg). The percentage of the undescribed compounds in the extract plant were: 12.5 ppm (compound 1), 150.0 ppm (compound 2), and 10.0 ppm (compound 3), respectively. The percentage of the undescribed compounds in the dry plant were: 1.9 ppm (compound 1), 23.1 ppm (compound 2), and 1.5 ppm (compound 3), respectively.
4.4. Pyruvate Dehydrogenase Complex (PDH) Cellular Activity
Point two percent gelatin was added to a black 96-well plate with a clear bottom and incubated for 1 h. Afterwards, the plate was washed with growth media. Human AD-293 cells, derived from the HEK293 cells, were seeded into black 96-well plates with a clear bottom and grown for 24 h. Compounds were then added and incubated for 24 h. The cells were then fixed with 2% paraformaldehyde, and permeabilized anti-PDHE1 pSer300 (Merk Millipore, Burlington, MA, AP1064) was added and incubated overnight. Next, the cells were washed and Alexa fluor 488, goat anti-rabbit ab (Invitrogen, Waltham, MA, A11008) was added with Hoechst 33,258 (Invitrogen, H3569) and incubated for 2 h. Finally, cells were washed, and the plates were measured in Operetta (PerkinElmer, Waltham, MA). The raw data were normalized for the pharmacological inhibitor control (5 mM dichloroacetate (DCA)) and percentage effect values using the software package Harmony High-Content Imaging and Analysis Software 3.1. Dose response curves were generated by plotting the percentage effect values and IC50 values were calculated via GraphPad Prism 6.
4.5. Statistical Analysis
All values are expressed as means ± standard error of the mean. The statistical significance threshold (p < 0.05 for all analyses) was assessed by one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons using Prism 5.01 software (GraphPad Software Inc., San Diego, CA, USA).
4.6. Spectroscopic Data
Compound
1. White amorphous powder;
: +31.2 (c 0.051, MeOH);
1H NMR (dimethyl sulfoxide-
d6, 600 MHz,) and
13C NMR data (dimethyl sulfoxide-
d6, 150 MHz), see
Table 1; HR-EI-MS
m/
z: 559.2159 [M + Na]
+ (calcd for C
27H
36O
11Na, 559.2155).
Compound
2. Colourless gum;
: +43.04 (c 0.049, MeOH);
1H NMR (acetone-
d6, 600 MHz,) and
13C NMR (acetone-
d6, 150 MHz), see
Table 1; HR-EI-MS
m/
z: 589.2264 [M + Na]
+ (calcd for C
28H
38O
12Na, 589.2261).
Compound
3. Colourless gum;
: −32.64 (c 0.053, MeOH);
1H NMR (methanol-
d4, 600 MHz,) and
13C NMR (methanol-
d4, 150 MHz), see
Table 1; HR-EI-MS
m/
z: 589.2275 [M + Na]
+ (calcd for C
28H
38O
12Na, 589.2261).


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}