
Differential Methylation Patterns in Apomictic vs. Sexual Genotypes of the Diplosporous Grass Eragrostis curvula

 , ,
, , 
 and
and
Abstract
1. Introduction
2. Results
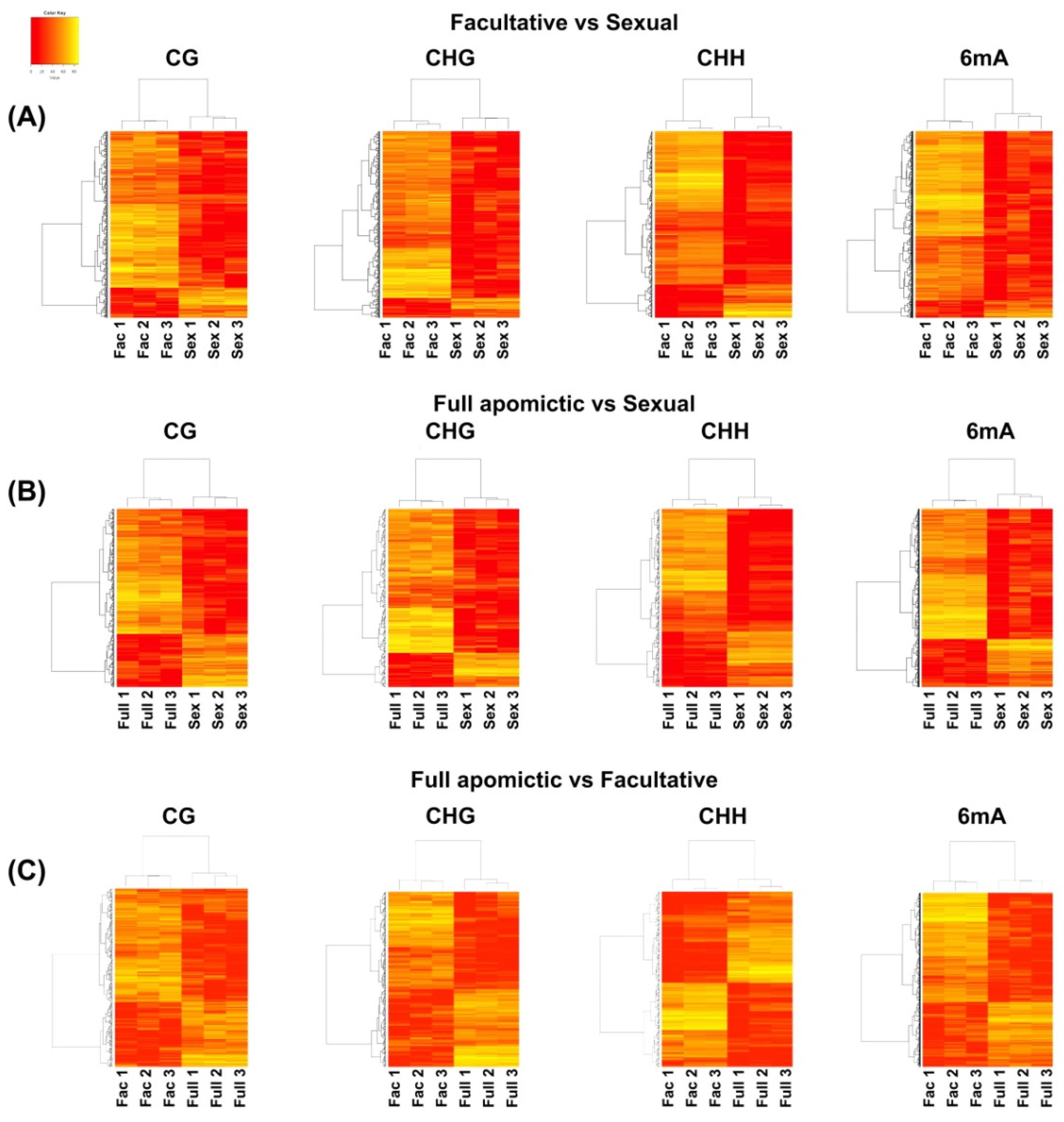
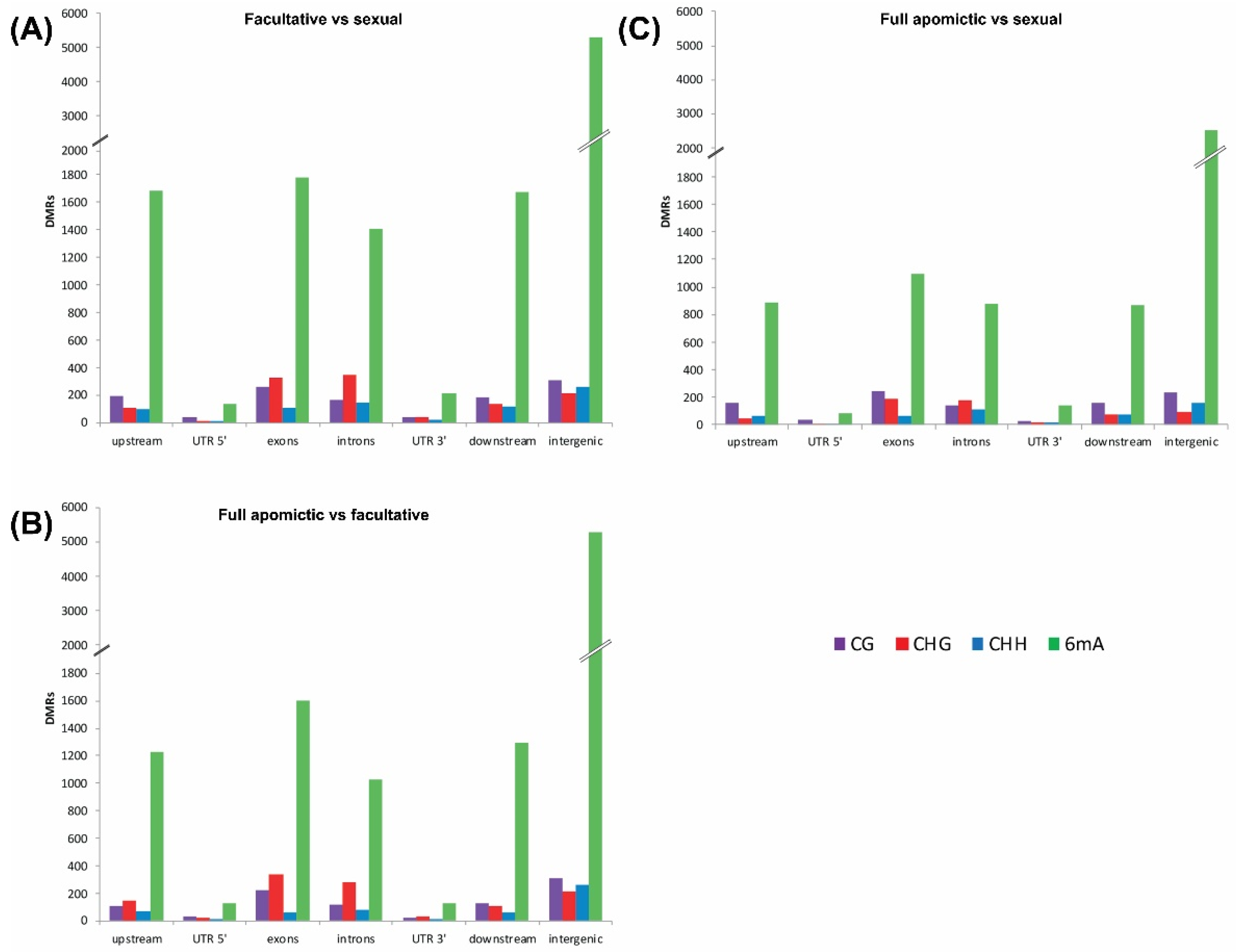
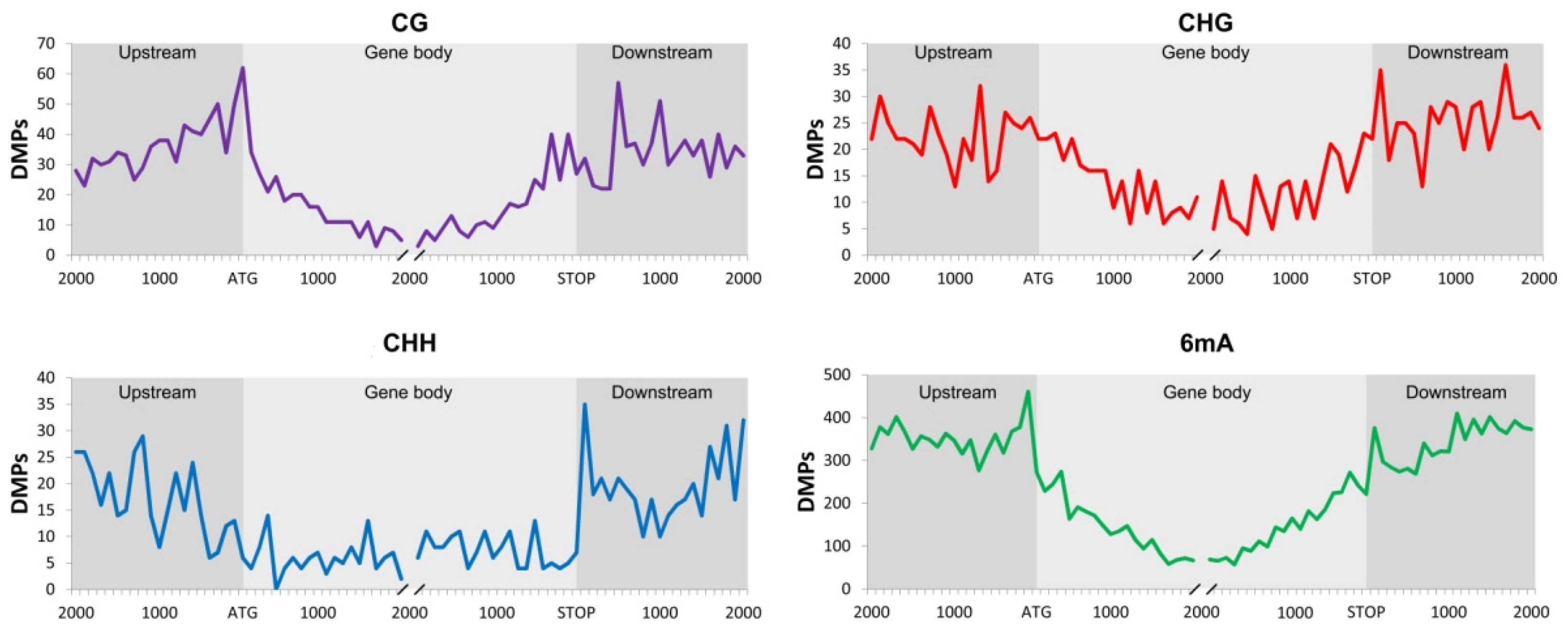
2.1. Differentially Methylated Positions (DMPs) and Differentially Methylated Regions (DMRs)
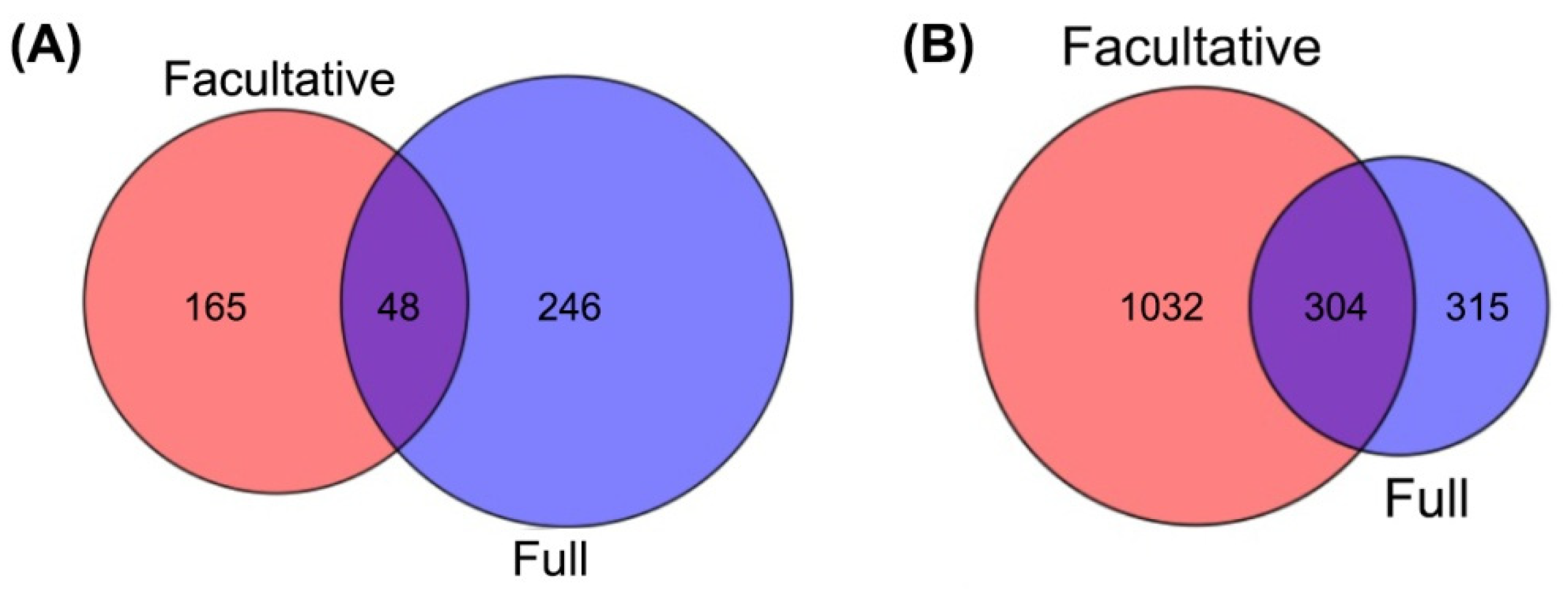
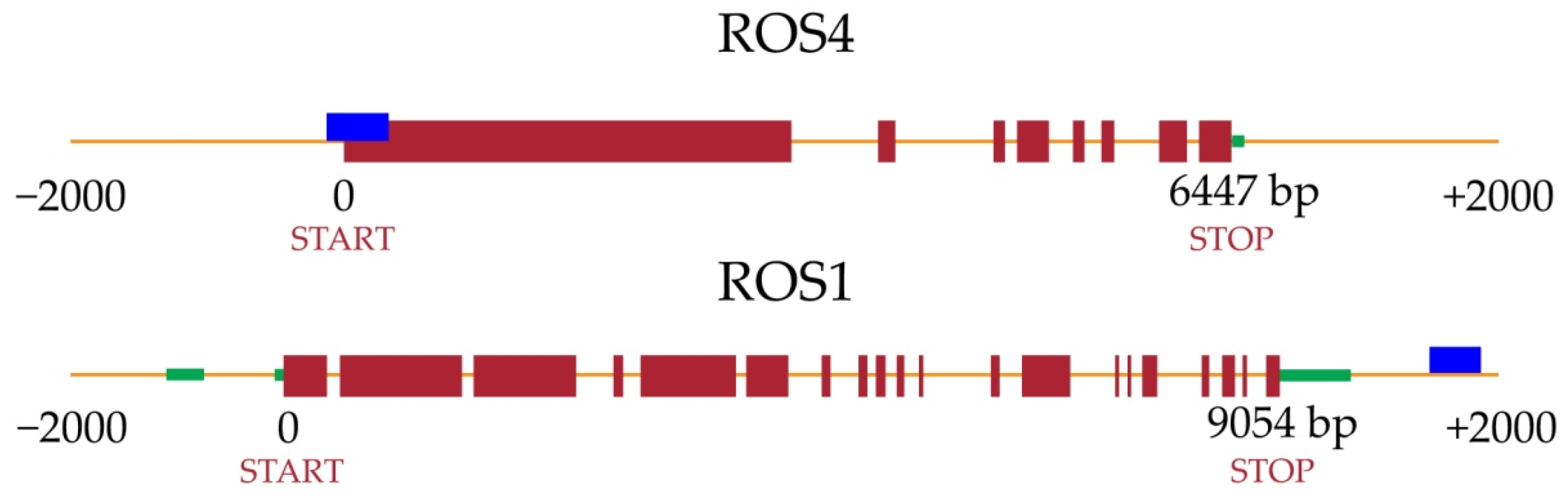
2.2. Identification of Differentially Methylated Genes (DMGs)
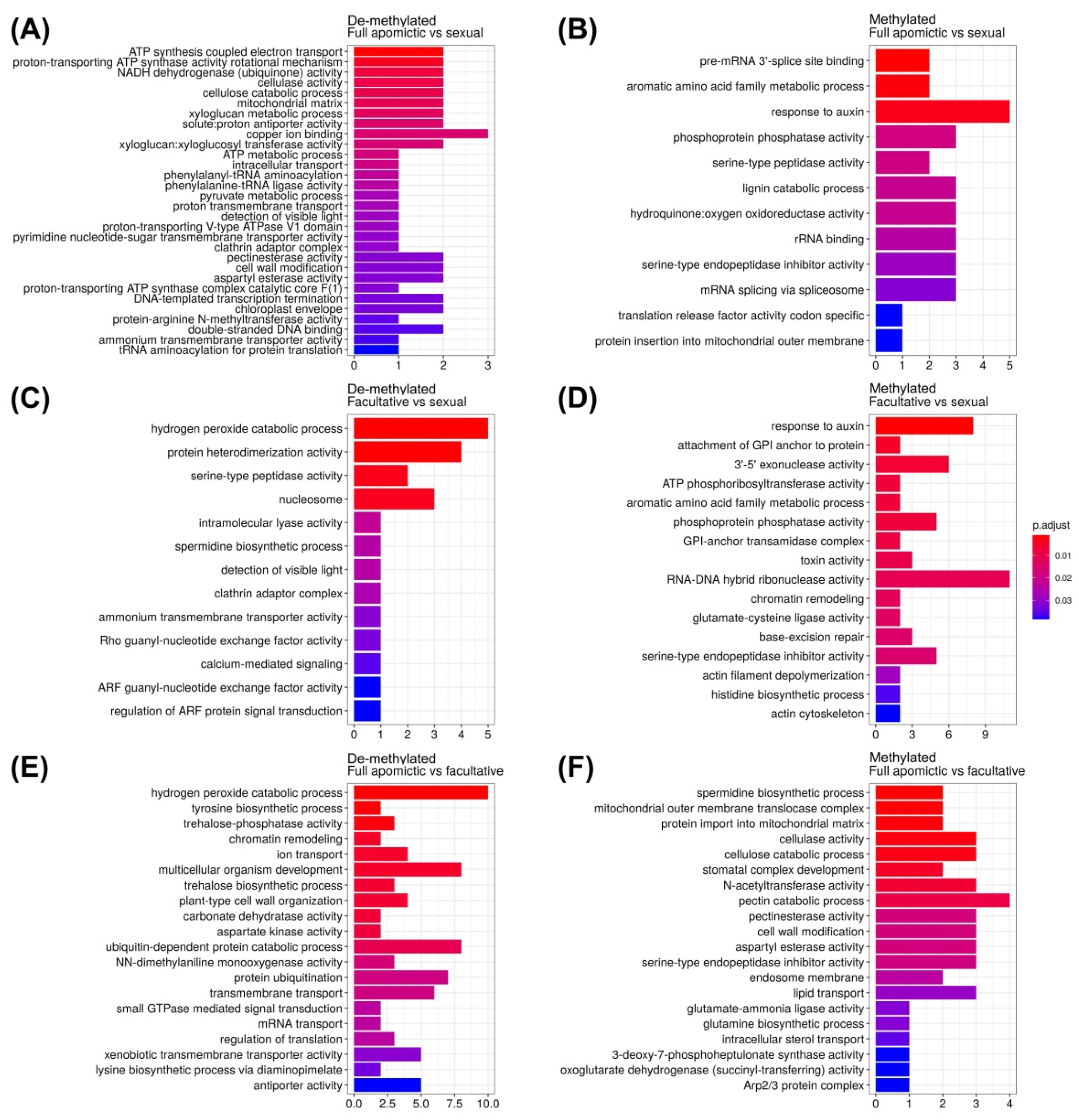
2.3. Gene Ontology Enrichment Analysis
2.4. Differentially Methylated Genes and RNA-Seq Expression
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. DNA Extraction
4.3. Library Construction and Sequencing (DNA Methylation Analysis)
4.4. Differentially Methylated Genes (DMGs)
4.5. Gene Ontology Analysis
4.6. Differentially Expressed Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Russo, V.E.; Martienssen, R.A.; Riggs, A.D. Epigenetic Mechanisms of Gene Regulation; Cold Spring Harbor Laboratory Press: Long Island, NY, USA, 1996. [Google Scholar]
- Chadwick, L.H. The NIH roadmap epigenomics program data resource. Epigenomics 2012, 4, 317–324. [Google Scholar] [CrossRef]
- Bräutigam, K.; Cronk, Q. DNA methylation and the evolution of developmental complexity in plants. Front. Plant Sci. 2018, 9, 1447. [Google Scholar] [CrossRef]
- Pikaard, C.S.; Scheid, O.M. Epigenetic regulation in plants. CSH Perspect. Biol. 2014, 6, 019315. [Google Scholar] [CrossRef]
- Sudan, J.; Raina, M.; Singh, R. Plant epigenetic mechanisms: Role in abiotic stress and their generational heritability. 3 Biotech. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lang, Z.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Bio. 2018, 19, 489–506. [Google Scholar] [CrossRef]
- Lang, Z.; Wang, Y.; Tang, K.; Tang, D.; Datsenka, T.; Cheng, J.; Zhang, Y.; Handa, A.K.; Zhu, J.K. Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. USA 2017, 114, E4511–E4519. [Google Scholar] [CrossRef]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc. Natl. Acad. Sci. USA 1996, 93, 8449–8454. [Google Scholar] [CrossRef] [PubMed]
- Madzima, T.F.; Mills, E.S.; Gardiner, J.M.; McGinnis, K.M. Identification of epigenetic regulators of a transcriptionally silenced transgene in maize. G3 2011, 1, 75–83. [Google Scholar] [CrossRef]
- Iwasaki, M.; Paszkowski, J. Identification of genes preventing transgenerational transmission of stress-induced epigenetic states. Proc. Natl. Acad. Sci. USA 2014, 111, 8547–8552. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef]
- Jones, L.; Ratcliff, F.; Baulcombe, D.C. RNA-directed transcriptional gene silencing in plants can be inherited independently of the RNA trigger and requires Met1 for maintenance. Curr. Biol. 2001, 11, 747–757. [Google Scholar] [CrossRef]
- Lindroth, A.M.; Cao, X.; Jackson, J.P.; Zilberman, D.; McCallum, C.M.; Henikoff, S.; Jacobsen, S.E. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 2001, 292, 2077–2080. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Aufsatz, W.; Zilberman, D.; Mette, M.F.; Huang, M.S.; Matzke, M.; Jacobsen, S.E. Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation. Curr. Biol. 2003, 13, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lim, J.Y.; Shin, H.; Kim, B.G.; Yoo, S.D.; Kim, W.T.; Huh, J.H. ROS1-dependent DNA demethylation is required for ABA-inducible NIC3 expression. Plant Physiol. 2019, 179, 1810–1821. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Kim, M.Y.; Vickers, M.; Park, J.S.; Hyun, Y.; Okamoto, T.; Zilberman, D.; Fischer, R.L.; Feng, X.; Choi, Y.; et al. DNA demethylation is initiated in the central cells of Arabidopsis and rice. Proc. Natl. Acad. Sci. USA 2016, 113, 15138–15143. [Google Scholar] [CrossRef]
- Ortega-Galisteo, A.P.; Morales-Ruiz, T.; Ariza, R.R.; Roldán-Arjona, T. Arabidopsis DEMETER-LIKE proteins DML2 and DML3 are required for appropriate distribution of DNA methylation marks. Plant Mol. Biol. 2008, 67, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S.; et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012, 13, 300. [Google Scholar] [CrossRef] [PubMed]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Do Kim, K.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Vanyushin, B.F.; Tkacheva, S.G.; Belozersky, A.N. Rare bases in animal DNA. Nature 1970, 225, 948–949. [Google Scholar] [CrossRef]
- Liang, Z.; Shen, L.; Cui, X.; Bao, S.; Geng, Y.; Yu, G.; Liang, F.; Xie, S.; Lu, T.; Gu, X.; et al. DNA N6-adenine methylation in Arabidopsis thaliana. Dev. Cell 2018, 45, 406–416. [Google Scholar] [CrossRef]
- Marconi, G.; Capomaccio, S.; Comino, C.; Acquadro, A.; Portis, E.; Porceddu, A.; Albertini, E. Methylation content sensitive enzyme ddRAD (MCSeEd): A reference-free, whole genome profiling system to address cytosine/adenine methylation changes. Sci. Rep. 2019, 9, 14864. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wang, C.; Liu, H.; Zhou, Q.; Liu, Q.; Guo, Y.; Peng, T.; Song, J.; Zhang, J.; Chen, L.; et al. Identification and analysis of adenine N 6-methylation sites in the rice genome. Nat. Plants 2018, 4, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Kurdyukov, S.; Bullock, M. DNA methylation analysis: Choosing the right method. Biology 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Vaisvila, R.; Ponnaluri, V.C.; Sun, Z.; Langhorst, B.W.; Saleh, L.; Guan, S.; Davis, T.B. EM-seq: Detection of DNA methyla-tion at single base resolution from picograms of DNA. BioRxiv 2020, 2019, 12. [Google Scholar] [CrossRef]
- Hand, M.L.; Koltunow, A.M. The genetic control of apomixis: Asexual seed formation. Genetics 2014, 197, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, D.; Johnson, S.D.; Henderson, S.T.; Koltunow, A.M. Genetic separation of autonomous endosperm formation (AutE) from the two other components of apomixis in Hieracium. Plant Reprod. 2013, 26, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A. Controlling apomixis: Shared features and distinct characteristics of gene regulation. Genes 2020, 11, 329. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.W.; Bashaw, E.C. Facultative apomixis in Eragrostis curvula. Crop. Sci. 1976, 16, 803–806. [Google Scholar] [CrossRef]
- Crane, C.F. Classification of apomictic mechanisms. In The Flowering of Apomixis: From Mechanisms to Genetic Engineering; Savidan, Y., Carman, J.G., Dresselhaus, T., Eds.; European Commission, ClMMYT, IRD: Veracruz Mexico, D.F., Mexico, 2001; pp. 24–43. [Google Scholar]
- Meier, M.; Zappacosta, D.; Selva, J.P.; Pessino, S.; Echenique, V. Evaluation of different methods for assessing the reproductive mode of weeping lovegrass plants, Eragrostis curvula (Schrad.) Nees. Aust. J. Bot. 2011, 59, 253–261. [Google Scholar] [CrossRef]
- Cervigni, G.D.; Paniego, N.; Díaz, M.; Selva, J.P.; Zappacosta, D.; Zanazzi, D.; Landerreche, I.; Martelotto, L.; Felitti, S.; Pessino, S.; et al. Expressed sequence tag analysis and development of gene associated markers in a near-isogenic plant system of Eragrostis curvula. Plant Mol. Biol. 2008, 67, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Garbus, I.; Romero, J.; Selva, J.P.; Pasten, M.C.; Chinestra, C.; Carballo, J.; Zappacosta, D.; Echenique, V. De novo transcriptome sequencing and assembly from apomictic and sexual Eragrostis curvula genotypes. PLoS ONE 2017, 12, e0185595. [Google Scholar] [CrossRef]
- Selva, J.P.; Zappacosta, D.; Carballo, J.; Rodrigo, J.M.; Bellido, A.; Gallo, C.A.; Gallardo, J.; Echenique, V. Genes modulating the increase in sexuality in the facultative diplosporous grass Eragrostis curvula under water stress conditions. Genes 2020, 11, 969. [Google Scholar] [CrossRef]
- Zappacosta, D.; Gallardo, J.; Carballo, J.; Meier, M.; Rodrigo, J.M.; Gallo, C.; Selva, J.P.; Stein, J.; Ortiz, J.P.; Albertini, E.; et al. A high-density linkage map of the forage grass Eragrostis curvula and localization of the diplospory locus. Front. Plant Sci. 2019, 10, 918. [Google Scholar] [CrossRef]
- Carballo, J.; Santos, B.; Zappacosta, D.; Garbus, I.; Selva, J.P.; Gallo, C.; Díaz, A.; Albertini, E.; Caccamo, M.; Echenique, V. A high-quality genome of Eragrostis curvula grass provides insights into Poaceae evolution and supports new strategies to enhance forage quality. Sci. Rep. 2019, 9, 10250. [Google Scholar] [CrossRef]
- Rodrigo, J.M.; Zappacosta, D.C.; Selva, J.P.; Garbus, I.; Albertini, E.; Echenique, V. Apomixis frequency under stress conditions in weeping lovegrass (Eragrostis curvula). PLoS ONE 2017, 12, 0175852. [Google Scholar] [CrossRef] [PubMed]
- Zappacosta, D.; Ochogavía, A.C.; Rodrigo, J.M.; Romero, J.R.; Meier, M.S.; Garbus, I.; Pessino, S.C.; Echenique, V.C. Increased apomixis expression concurrent with genetic and epigenetic variation in a newly synthesized Eragrostis curvula polyploid. Sci. Rep. 2014, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Selva, J.P.; Siena, L.; Rodrigo, J.M.; Garbus, I.; Zappacosta, D.; Romero, J.R.; Ortiz, J.P.; Pessino, S.C.; Leblanc, O.; Echenique, V. Temporal and spatial expression of genes involved in DNA methylation during reproductive development of sexual and apomictic Eragrostis curvula. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Garbus, I.; Selva, J.P.; Pasten, M.C.; Bellido, A.M.; Carballo, J.; Albertini, E.; Echenique, V. Characterization and discovery of miRNA and miRNA targets from apomictic and sexual genotypes of Eragrostis curvula. BMC Genom. 2019, 20, 839. [Google Scholar] [CrossRef]
- Podio, M.; Cáceres, M.E.; Samoluk, S.S.; Seijo, J.G.; Pessino, S.C.; Ortiz, J.P.A.; Pupilli, F. A methylation status analysis of the apomixis-specific region in Paspalum spp. suggests an epigenetic control of parthenogenesis. J. Exp. Bot. 2014, 65, 6411–6424. [Google Scholar] [CrossRef]
- Garcia-Aguilar, M.; Michaud, C.; Leblanc, O.; Grimanelli, D. Inactivation of a DNA methylation pathway in maize reproductive organs results in apomixis-like phenotypes. Plant Cell 2010, 22, 3249–3267. [Google Scholar] [CrossRef]
- Olmedo-Monfil, V.; Durán-Figueroa, N.; Arteaga-Vandázquez, M.; Demesa-Arévalo, E.; Autran, D.; Grimanelli, D.; Grimanelli, D.; Slotkin, K.; Martienssen, R.A.; Vielle-Calzada, J.-P. Control of female gamete formation by a small RNA pathway in Arabidopsis. Nature 2010, 464, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Goel, S.; Meeley, R.B.; Dantec, C.; Parrinello, H.; Michaud, C.; Leblanc, O.; Grimanelli, D. Production of viable gametes without meiosis in maize deficient for an ARGONAUTE protein. Plant Cell 2011, 23, 443–458. [Google Scholar] [CrossRef]
- Hernández-Lagana, E.; Rodríguez-Leal, D.; Lúa, J.; Vielle-Calzada, J.P. A Multigenic network of ARGONAUTE4 clade members controls early megaspore formation in Arabidopsis. Genetics 2016, 204, 1045–1056. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Melnick, A.; Mason, C.E. MethylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.Q.; Zhang, Y.J.; Zhou, S.R.; Hu, W.Y.; Wu, X.T.; Ye, Y.J.; Wu, X.X.; Xiao, Y.P.; Li, X.; Xue, H.W. Global analysis reveals the crucial roles of DNA methylation during rice seed development. Plant Physiol. 2015, 168, 1417–1432. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Lyu, J.; Li, Q.; Liu, H.; Wang, D.; Zhang, M.; Springer, N.M.; Ross-Ibarra, J.; Yang, J. Evolutionary and functional genomics of DNA methylation in maize domestication and improvement. Nat. Commun. 2020, 11, 5539. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.G.; Wang, X.; Zhang, L.; Xiong, X.; Zhang, Z.; Tang, K.; Pan, L.; Hsu, C.C.; Xu, H.; Tao, W.A.; et al. A protein complex regulates RNA processing of intronic heterochromatin-containing genes in Arabidopsis. Proc. Natl. Acad. Sci. USA 2017, 114, E7377–E7384. [Google Scholar] [CrossRef] [PubMed]
- Zühl, L.; Volkert, C.; Ibberson, D.; Schmidt, A. Differential activity of F-box genes and E3 ligases distinguishes sexual versus apomictic germline specification in Boechera. J. Exp. Bot. 2019, 70, 5643–5657. [Google Scholar] [CrossRef] [PubMed]
- Bocchini, M.; Galla, G.; Pupilli, F.; Bellucci, M.; Barcaccia, G.; Ortiz, J.P.; Pessino, S.C.; Albertini, E. The vesicle trafficking regulator PN_SCD1 is demethylated and overexpressed in florets of apomictic Paspalum notatum genotypes. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Okada, T.; Hu, Y.; Tucker, M.R.; Taylor, J.M.; Johnson, S.D.; Spriggs, A.; Tsuchiya, T.; Oelkers, K.; Rodrigues, J.C.; Koltunow, A.M. Enlarging cells initiating apomixis in Hieracium praealtum transition to an embryo sac program prior to entering mitosis. Plant Physiol. 2013, 163, 216–231. [Google Scholar] [CrossRef]
- Galla, G.; Zenoni, S.; Avesani, L.; Altschmied, L.; Rizzo, P.; Sharbel, T.F.; Barcaccia, G. Pistil transcriptome analysis to disclose genes and gene products related to aposporous apomixis in Hypericum perforatum L. Front. Plant Sci. 2017, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Lechner, E.; Leonhardt, N.; Eisler, H.; Parmentier, Y.; Alioua, M.; Jacquet, H.; Leung, J.; Genschik, P. MATH/BTB CRL3 receptors tar-get the homeodomain-leucine zipper ATHB6 to modulate abscisic acid signaling. Dev. Cell 2011, 21, 1116–1128. [Google Scholar] [CrossRef]
- Weber, H.; Hellmann, H. Arabidopsis thaliana BTB/POZ-MATH proteins interact with members of the ERF/AP2 transcription factor family. FEBS J. 2009, 276, 6624–6635. [Google Scholar] [CrossRef]
- Li, X.; Qian, W.; Zhao, Y.; Wang, C.; Shen, J.; Zhu, J.K.; Gong, Z. Antisilencing role of the RNA-directed DNA methylation pathway and a histone acetyltransferase in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, 11425–11430. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Miki, D.; Zhang, H.; Liu, Y.; Zhang, X.; Tang, K.; Kan, Y.; La, H.; Li, X.; Li, S.; et al. A histone acetyltransferase regulates active DNA demethylation in Arabidopsis. Science 2012, 336, 1445–1448. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Zhang, H.; Julian, R.; Tang, K.; Xie, S.; Zhu, J.K. Regulatory link between DNA methylation and active demethylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, 3553–3557. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.P.; Pignatta, D.; Henikoff, S.; Gehring, M. Methylation-sensitive expression of a DNA demethylase gene serves as an epigenetic rheostat. PLoS Gen. 2015, 11, e1005142. [Google Scholar] [CrossRef]
- Xie, Y.; Zhou, Q.; Zhao, Y.; Li, Q.; Liu, Y.; Ma, M.; Wang, B.; Shen, R.; Zheng, Z.; Wang, H. FHY3 and FAR1 integrate light signals with the miR156-SPL module-mediated aging pathway to regulate Arabidopsis flowering. Mol. Plant 2020, 13, 483–498. [Google Scholar] [CrossRef]
- Niu, B.; Wang, L.; Zhang, L.; Ren, D.; Ren, R.; Copenhaver, G.P.; Ma, H.; Wang, Y. Arabidopsis cell division cycle 20.1 is required for normal meiotic spindle assembly and chromosome segregation. Plant Cell 2015, 27, 3367–3382. [Google Scholar] [CrossRef]
- Khanday, I.; Skinner, D.; Yang, B.; Mercier, R.; Sundaresan, V. A male-expressed rice embryogenic trigger redirected for asexual propagation through seeds. Nature 2019, 565, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, D.D.; Köhler, C. Auxin: A molecular trigger of seed development. Genes Dev. 2018, 32, 479–490. [Google Scholar] [CrossRef]
- Pagnussat, G.C.; Alandete-Saez, M.; Bowman, J.L.; Sundaresan, V. Auxin-dependent patterning and gamete specification in the Arabidopsis female gametophyte. Science 2009, 324, 1684–1689. [Google Scholar] [CrossRef]
- Khanday, I.; Sundaresan, V. Plant zygote development: Recent insights and applications to clonal seeds. Curr. Opin. Plant Biol. 2021, 59, 101993. [Google Scholar] [CrossRef] [PubMed]
- McMichael, C.M.; Reynolds, G.D.; Koch, L.M.; Wang, C.; Jiang, N.; Nadeau, J.; Sack, F.D.; Gelderman, M.B.; Pan, J.; Bednarek, S.Y. Mediation of clathrin-dependent trafficking during cytokinesis and cell expansion by Arabidopsis stomatal cytokinesis defective proteins. Plant Cell 2013, 25, 3910–3925. [Google Scholar] [CrossRef] [PubMed]
- Ranocha, P.; Dima, O.; Nagy, R.; Felten, J.; Corratgé-Faillie, C.; Novák, O.; Morreel, K.; Lacombe, B.; Martinez, Y.; Pfrunder, S.; et al. Arabidopsis WAT1 is a vacuolar auxin transport facilitator required for auxin homoeostasis. Nat. Commun. 2013, 16, 2625. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Li, G.; Qu, D.; Li, X.; Wang, Y. Into the seed: Auxin controls seed development and grain yield. Int. J. Mol. Sci. 2020, 21, 1662. [Google Scholar] [CrossRef]
- Di Marsico, M.; Cerruti, E.; Comino, C.; Porceddu, A.; Acquadro, A.; Capomaccio, S.; Marconi, G.; Albertini, E. MCSeEd (Methylation Context Sensitive Enzyme ddRAD): A New Method to Analyze DNA Methylation. In Plant Epigenetics and Epigenomics Methods in Molecular Biology; Spillane, C., McKeown, P., Eds.; Humana: New York, NY, USA, 2020; pp. 47–64. [Google Scholar]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Lawrence, M.; Huber, W.; Pages, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upstream | Gene Body | Downstream | |||||
|---|---|---|---|---|---|---|---|
| Meth | De-meth | Meth | De-meth | Meth | De-meth | ||
| Context | DMRs | Facultative vs. Sexual | |||||
| CG | 448 | 160 | 29 | 139 | 46 | 141 | 31 |
| CHG | 459 | 98 | 16 | 214 | 29 | 117 | 22 |
| CHH | 383 | 76 | 16 | 110 | 13 | 89 | 20 |
| 6mA | 7343 | 1473 | 191 | 1659 | 198 | 1484 | 159 |
| Full Apomictic vs. Sexual | |||||||
| CG | 362 | 108 | 55 | 94 | 63 | 109 | 44 |
| CHG | 233 | 35 | 10 | 113 | 25 | 53 | 18 |
| CHH | 251 | 36 | 26 | 66 | 21 | 55 | 21 |
| 6mA | 3862 | 625 | 260 | 875 | 305 | 629 | 250 |
| Full Apomictic vs. Facultative | |||||||
| CG | 307 | 44 | 72 | 49 | 98 | 39 | 66 |
| CHG | 368 | 85 | 113 | 102 | 154 | 71 | 113 |
| CHH | 232 | 22 | 35 | 35 | 41 | 33 | 34 |
| 6mA | 5070 | 462 | 821 | 543 | 1049 | 438 | 734 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carballo, J.; Zappacosta, D.; Marconi, G.; Gallardo, J.; Di Marsico, M.; Gallo, C.A.; Caccamo, M.; Albertini, E.; Echenique, V. Differential Methylation Patterns in Apomictic vs. Sexual Genotypes of the Diplosporous Grass Eragrostis curvula. Plants 2021, 10, 946. https://doi.org/10.3390/plants10050946
Carballo J, Zappacosta D, Marconi G, Gallardo J, Di Marsico M, Gallo CA, Caccamo M, Albertini E, Echenique V. Differential Methylation Patterns in Apomictic vs. Sexual Genotypes of the Diplosporous Grass Eragrostis curvula. Plants. 2021; 10(5):946. https://doi.org/10.3390/plants10050946
Chicago/Turabian StyleCarballo, Jose, Diego Zappacosta, Gianpiero Marconi, Jimena Gallardo, Marco Di Marsico, Cristian A. Gallo, Mario Caccamo, Emidio Albertini, and Viviana Echenique. 2021. "Differential Methylation Patterns in Apomictic vs. Sexual Genotypes of the Diplosporous Grass Eragrostis curvula" Plants 10, no. 5: 946. https://doi.org/10.3390/plants10050946
APA StyleCarballo, J., Zappacosta, D., Marconi, G., Gallardo, J., Di Marsico, M., Gallo, C. A., Caccamo, M., Albertini, E., & Echenique, V. (2021). Differential Methylation Patterns in Apomictic vs. Sexual Genotypes of the Diplosporous Grass Eragrostis curvula. Plants, 10(5), 946. https://doi.org/10.3390/plants10050946