
Molecular Characterization of the Coat Protein Gene of Greek Apple Stem Pitting Virus Isolates: Evolution through Deletions, Insertions, and Recombination Events

 ,
,  , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Results
2.1. Development of a Specific RT–PCR Assay for ASPV Detection
2.2. Detection and Incidence of ASPV in Quince in Greece
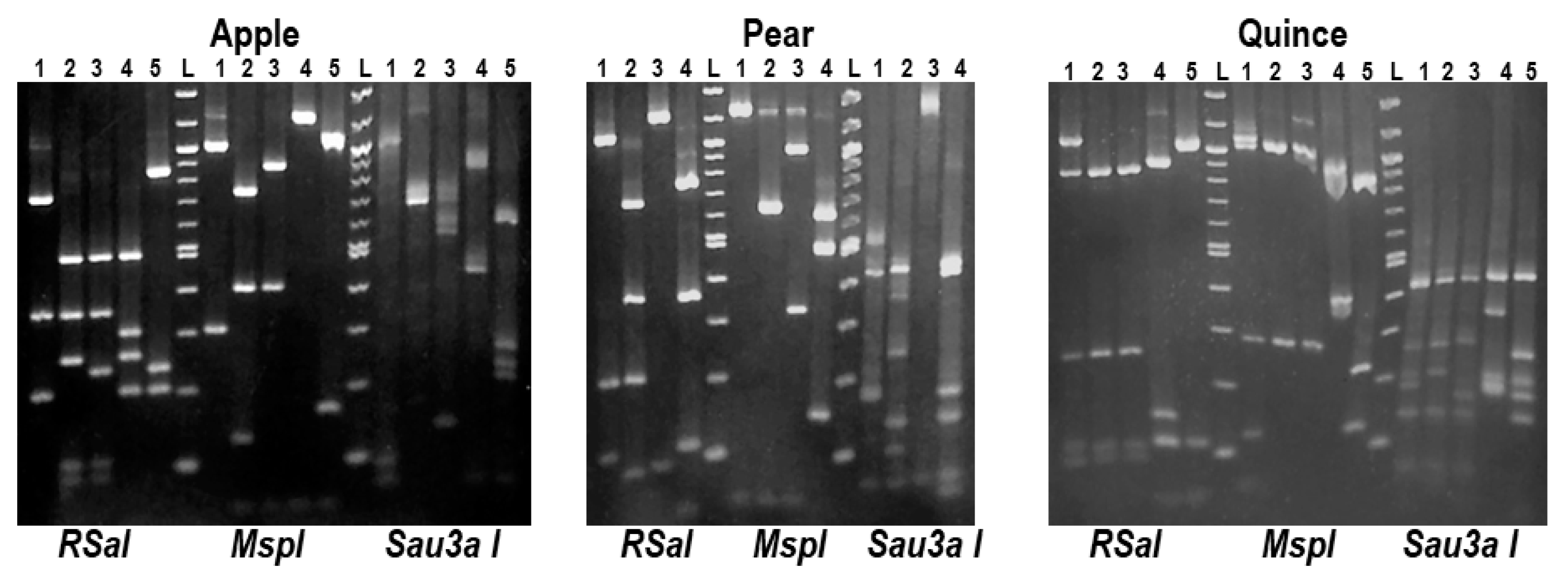
2.3. Preliminary Analysis for the Presence of ASPV Variants by RFLP
2.4. Molecular Characterization of the Complete CP and Partial 3΄UTR Sequences of the 14 Selected Greek Isolates
2.5. Molecular Variability Analysis of ASPV Isolates
2.6. Recombination Analysis
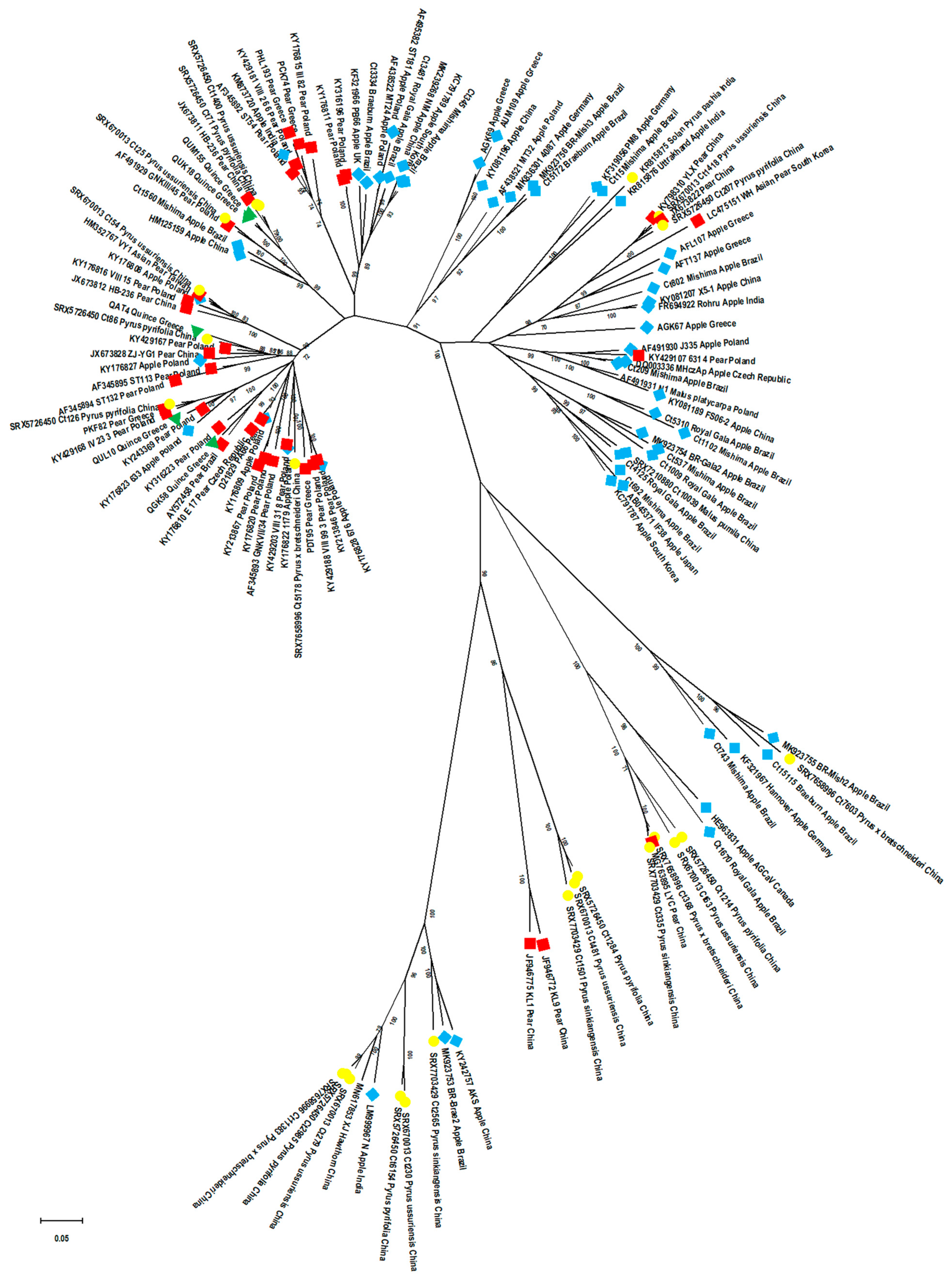
2.7. Phylogenetic Relationships among ASPV Isolates: Potential Effect of CP Size, Host and Country of Origin, and of Virus Detection Method
2.8. Evolutionary Networks
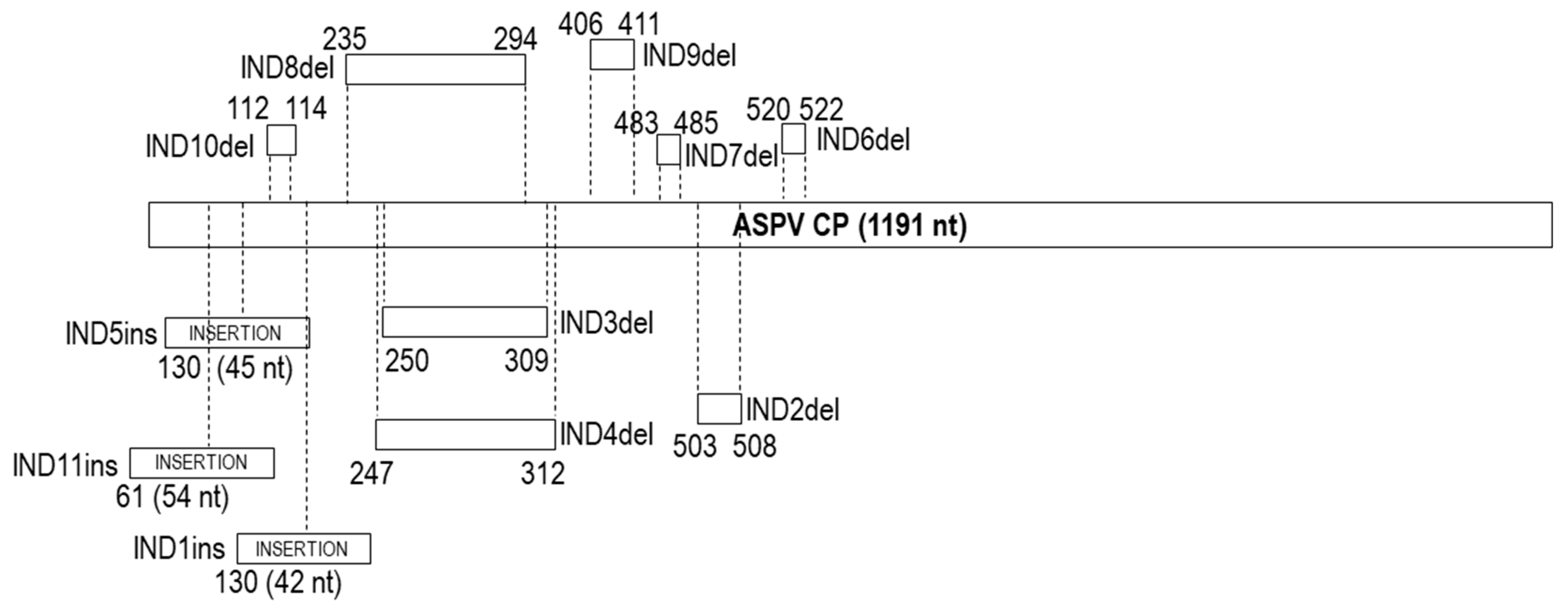
2.9. Characterization of Insertions/Deletions (Indels): Type, Position, Variability and Their Impact on Population Structure
2.10. Analysis of Selection Pressure in the ASPV CP Gene
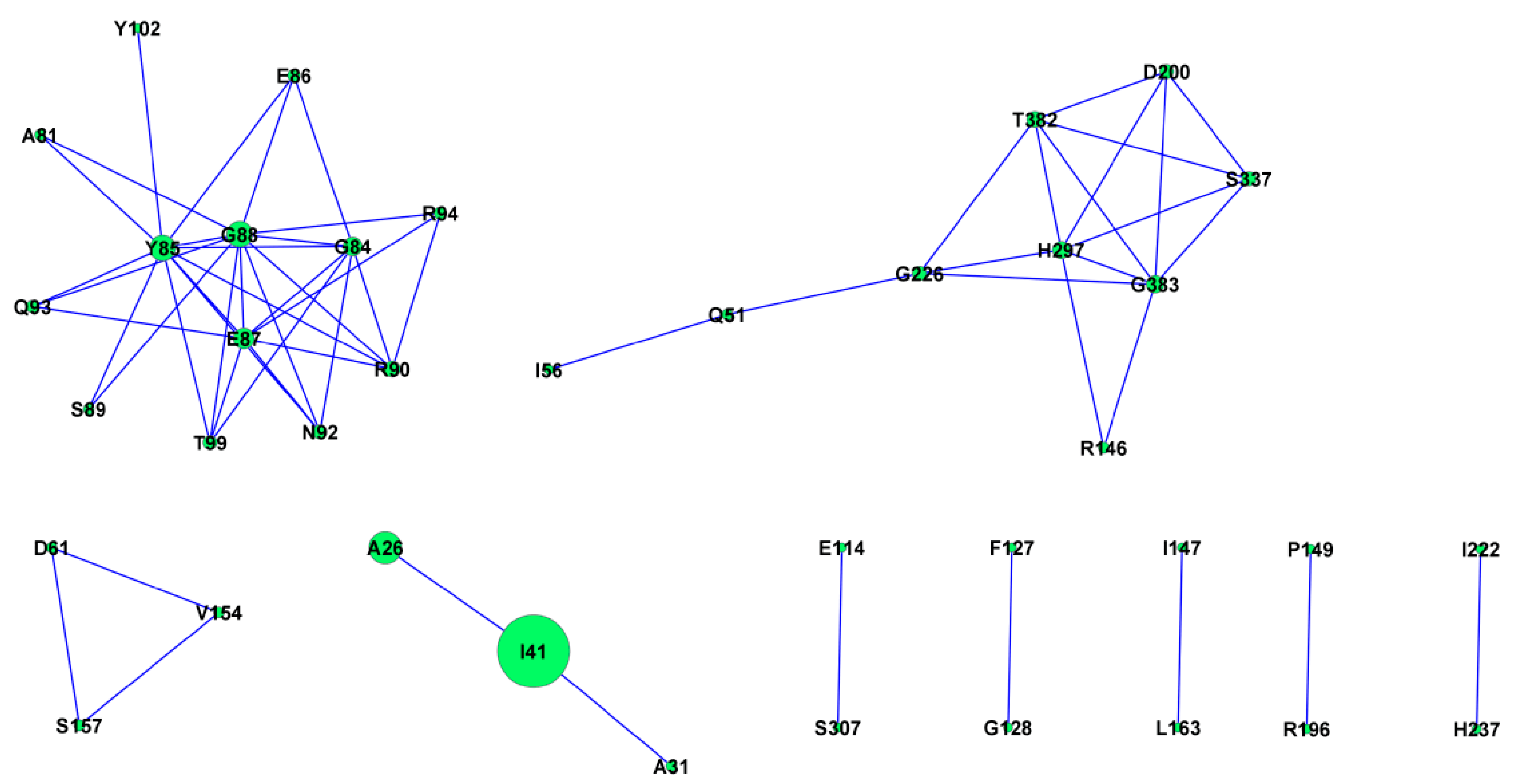
2.11. Protein Co-Evolution Networks of ASPV CP
3. Discussion
4. Materials and Methods
4.1. Viral Isolates and Development of a Two-Step RT–PCR Assay for ASPV Detection
4.2. Restriction Fragment Length Polymorphism (RFLP) Analysis
4.3. Cloning and Sequence Analysis
4.4. Identification of Additional ASPV CP Gene Sequences from High-Throughput Sequence Data
4.5. Molecular Variability and Recombination Analysis
4.6. Phylogenetic Analysis and Evolutionary Networks
4.7. Inference of Selection Pressure in the ASPV CP Gene and Co-Evolution of Amino Acids
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nemeth, M. Virus, Mycoplasma and Ricketsia Diseases of Fruit Trees; Academiai Kiado: Budapest, Hungary, 1986. [Google Scholar]
- Jelkmann, W. Nucleotide sequences of apple stem pitting virus and of the coat protein gene of a similar virus from pear associated with vein yellows disease and their relationship with potex- and carlaviruses. J. Gen. Virol. 1994, 75, 1535–1545. [Google Scholar] [CrossRef]
- Adams, M.J.; Antoniw, J.F.; Bar-Joseph, M.; Brunt, A.A.; Candresse, T.; Foster, G.D.; Martelli, G.P.; Milne, R.G.; Zavriev, S.K.; Fauquet, C.M. The new plant virus family Flexiviridae and assessment of molecular criteria for species demarcation. Arch. Virol. 2004, 149, 1045–1060. [Google Scholar] [CrossRef]
- Fauquet, C.M.; Mayo, M.A.; Maniloff, J.; Desselberger, U.; Ball, L.A. Virus Taxonomy. Eighth Report of the International Committee on Taxonomy of Viruses; Academic Press: New York, NY, USA, 2005. [Google Scholar]
- Mathioudakis, M.M.; Maliogka, V.I.; Dovas, C.I.; Vasilakakis, M.; Katis, N.I. First record of the apple stem pitting virus (ASPV) in quince in Greece. J. Plant Pathol. 2006, 88, 221. [Google Scholar]
- Mathioudakis, M.M.; Maliogka, V.I.; Dovas, C.I.; Paunovic, S.; Katis, N.I. Reliable RT-PCR detection of apple stem pitting virus in pome fruits ant its association with quince fruit deformation disease. Plant Pathol. 2009, 58, 228–236. [Google Scholar] [CrossRef]
- Stouffer, R.F. Apple stem pitting. In Virus and Virus-like Diseases of Pome Fruits and Simulating Noninfectious Disorders; Fridlund, P.R., Ed.; Washington State University Cooperative Extension College: Pullman, WA, USA, 1989; pp. 138–144. [Google Scholar]
- Leone, G.; Linder, J.L.; Van der Meer, F.A.; Schoen, C.D. Symptoms on apple and pear indicators after back-transmission from Nicotiana occidentalis confirm the identity of apple stem pitting virus with pear vein yellows virus. Acta Hort. 1998, 472, 61–65. [Google Scholar] [CrossRef]
- James, D.; Varga, A.; Jesperson, G.D.; Navratil, M.; Safarova, D.; Constable, F.; Horner, M.; Eastwell, K.; Jelkmann, W. Identification and complete genome analysis of a virus variant or putative new foveavirus associated with apple green crinkle disease. Arch. Virol. 2013, 158, 1877–1887. [Google Scholar] [CrossRef]
- Morelli, M.; Giampetruzzi, A.; Laghezza, L.; Catalano, L.; Savino, V.N.; Saldarelli, P. Identification and characterization of an isolate of apple green crinkle associated virus involved in a severe disease of quince (Cydonia oblonga, Mill.). Arch. Virol. 2017, 162, 299–306. [Google Scholar] [CrossRef]
- Constable, F.E.; Joyce, P.A.; Rodoni, B.C. A survey of key Australian pome fruit growing districts for exotic and endemic pathogens. Australas. Plant Path. 2007, 36, 165–172. [Google Scholar] [CrossRef]
- Ma, B.G.; Niu, J.X.; Morley-Bunker, M.; Pan, L.Z.; Zhang, H.P.; Zhang, L.X. Detection of three pear viruses by multiplex RT-PCR assays with co-amplification of an internal control. Australas. Plant Path. 2008, 37, 117–122. [Google Scholar] [CrossRef]
- Mathioudakis, M.M.; Maliogka, V.I.; Katsiani, A.T.; Katis, N.I. Incidence and molecular variability of apple stem pitting and apple chlorotic leaf spot viruses in apple and pear orchards in Greece. J. Plant Pathol. 2010, 92, 141–149. [Google Scholar]
- Mathioudakis, M.M.; Candresse, T.; Barone, M.; Rogozzino, A.; Katis, N.I. Cydonia japonica, Pyrus calleryana and P. amygdaliformis: Three new ornamental or wild hosts of apple stem pitting virus. Virus Genes 2011, 44, 319–322. [Google Scholar] [CrossRef]
- Dhir, S.; Tomar, M.; Thakur, P.D.; Ram, R.; Hallan, V.; Zaidi, A.A. Molecular evidence for apple stem pitting virus infection in India. Plant Pathol. 2010, 59, 393. [Google Scholar] [CrossRef]
- Yang, H.Y.; Liu, Z.J.; Luo, S.; Li, L.L. First report of apple stem pitting virus infecting Nanking cherry in China. Plant Dis. 2017, 101, 1067. [Google Scholar] [CrossRef]
- Morán, F.; Canales, C.; Olmos, A.; Ruiz-García, A.B. Loquat (Eriobotrya japonica) is a new natural host of apple stem pitting virus. Plants 2020, 9, 1560. [Google Scholar] [CrossRef]
- Matsuda, H.; Oda, Y.; Isogai, M.; Takahashi, T.; Ito, T.; Yoshida, K.; Yoshikawa, N. Genome heterogeneity of apple stem pitting virus in apple trees. Acta Hort. 2001, 550, 285–290. [Google Scholar]
- Gadiou, S.; Kundu, J.K.; Paunovic, S.; Garcia-Diez, P.; Komorowska, B.; Gospodaryk, A.; Handa, A.; Massart, S.; Birisik, N.; Takur, P.D.; et al. Genetic diversity of flexiviruses infecting pome fruit trees. J. Plant Pathol. 2010, 92, 685–691. [Google Scholar]
- Komorowska, B.; Siedlecki, P.; Kaczanowski, S.; Hasiów-Jaroszewska, B.; Malinowski, T. Sequence diversity and potential recombination events in the coat protein gene of apple stem pitting virus. Virus Res. 2011, 158, 263–267. [Google Scholar] [CrossRef]
- Liu, N.; Niu, J.; Zhao, Y. Complete genomic sequence analyses of apple stem pitting virus isolates from China. Arch. Virol. 2012, 44, 124–130. [Google Scholar] [CrossRef]
- Yoon, J.Y.; Joa, J.H.; San Choi, K.; Do, K.S.; Lim, H.C.; Chung, B.N. Genetic diversity of a natural population of apple stem pitting virus isolated from apple in Korea. Plant Pathol. J. 2014, 30, 195–199. [Google Scholar] [CrossRef]
- Ma, X.; Hong, N.; Moffett, P.; Wang, G. Genetic diversity and evolution of apple stem pitting virus isolates from pear in China. Canadian J. Plant Pathol. 2016, 38, 218–230. [Google Scholar] [CrossRef]
- Komorowska, B.; Hasiów-Jaroszewska, B.; Elena, S.F. Evolving by deleting: Patterns of molecular evolution of apple stem pitting virus isolates from Poland. J. Gen. Virol. 2019, 100, 1442–1456. [Google Scholar] [CrossRef]
- Jelkmann, W.; Keim-Konrad, R. Immuno-capture polymerase chain reaction and plate-trapped ELISA for the detection of apple stem pitting virus. J. Phytopathol. 1997, 145, 499–503. [Google Scholar] [CrossRef]
- Chooi, K.M.; Cohen, D.; Pearson, M.N. Molecular characterization of two divergent variants of grapevine leafroll-associated virus 3 in New Zealand. Arch. Virol. 2013, 158, 1597–1602. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.D. Recombination in Plant RNA Viruses. In Plant Virus Evolution; Roossinck, M.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Dhir, S.; Ram, R.; Hallan, V.; Zaidi, A.A. Molecular characterization of an Indian variant of apple stem pitting virus: Evidence of recombination. J. Plant Pathol. 2011, 93, 471–478. [Google Scholar]
- Li, Z.; Kondo, H.; Andika, I.B.; Liu, P.; Sun, L.; Wu, Y. Identification of genome recombination among apple stem pitting isolates. J. Plant Pathol. 2016, 98, 595–601. [Google Scholar]
- Adams, M.J.; Candresse, T.; Hammond, J.; Kreuze, J.F.; Martelli, G.P.; Namba, S.; Pearson, M.N.; Ryu, K.H.; Saldarelli, P.; Yoshikawa, N. Family Betafexiviridae. In Virus taxonomy. 9th Report of the ICTV; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2012; pp. 920–941. [Google Scholar]
- Chare, E.R.; Holmes, E.C. Selection pressures in the capsid genes of plant RNA viruses reflect mode of transmission. J. Gen. Virol. 2004, 85, 3149–3157. [Google Scholar] [CrossRef] [PubMed]
- Moury, B. Differential selection of genes of cucumber mosaic virus subgroups. Mol. Biol. Evol. 2004, 21, 1602–1611. [Google Scholar] [CrossRef] [PubMed]
- Moury, B.; Simon, V. dN/dS-based methods detect positive selection linked to trade-offs between different fitness traits in the coat protein of potato virus Y. Mol. Biol. Evol. 2011, 28, 2707–2717. [Google Scholar] [CrossRef]
- Fares, M.A.; Travers, S.A. A novel method for detecting intramolecular coevolution: Adding a further dimension to selective constraints analyses. Genetics 2006, 173, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kapralov, M.V.; Anisimova, M. Coevolution of amino acid residues in the key photosynthetic enzyme Rubisco. BMC Evol. Biol. 2010, 11, 266. [Google Scholar] [CrossRef]
- Komorowska, B.; Malinowski, T.; Zawadzka, B. Amplification of the coat protein gene of apple stem pitting virus (ASPV). Phytopathol. Pol. 1999, 17, 49–59. [Google Scholar]
- Caetano-Anolles, G. Amplifying DNA with arbitrary pligonucleotide primers. Genome Res. 1993, 3, 85–94. [Google Scholar] [CrossRef]
- Komorowska, B.; Malinowski, T.; Michalczuk, L. Evaluation of several RT-PCR primer pairs for the detection of apple stem pitting virus. J. Virol. Methods 2010, 168, 242–247. [Google Scholar] [CrossRef]
- Hu, G.J.; Dong, Y.F.; Zhang, Z.P.; Fan, X.; Ren, F.; Li, Z. Occurrence and genetic diversity analysis of apple stem pitting virus isolated from apples in China. Arch. Virol. 2017, 162, 2397–2402. [Google Scholar] [CrossRef]
- Ma, X.; Hong, N.; Moffett, P.; Zhou, Y.; Wang, G. Functional analysis of apple stem pitting virus coat protein variants. Virol. J. 2019, 16, 20. [Google Scholar] [CrossRef]
- Spiegel, S.; Thompson, D.; Varga, A.; Rosner, A.; James, D. An apple chlorotic leaf spot virus isolate from ornamental dwarf flowering almond (Prunus glandulosa ‘Sinensis’): Detection and characterization. Hortscience 2005, 40, 1401–1404. [Google Scholar] [CrossRef]
- Nickel, O.; Fajardo, T.V.M.; Candresse, T. First report on detection of three Bunya-like viruses in apples in Brazil. Plant Dis. 2020, 104, 3088. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The Clustal X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 24, 4876–4882. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 1–5. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. CABIOS 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, S. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Fares, M.A.; McNally, D. CAPS: Coevolution analysis using protein sequences. Bioinformatics 2006, 22, 2821–2822. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Country | Host | Recombinant Isolate | Major Parent (Host, Country) | Minor Parent (Host, Country) | No. of Methods with P < 0.05 |
|---|---|---|---|---|---|
| Greek | Quince | QUL10 | KY243369 (pear, Poland) | KY176823 (apple, Poland) | 6 |
| Pear | PCK74 | KY429181 (pear, Poland) | KY176815 (pear, Poland) | 7 | |
| Pear | PHL193 | KY429181 (pear, Poland) | KY176815 (pear, Poland) | 7 | |
| Botanical -China | P. ussuriensis | Ct54 (SRX670013) | JX673812 (pear, China) | JX673811 (pear, China) | 7 |
| Brazilian | Apple cv Mishima | Ct15 | KR815875 (pear, India) | AF345892 (pear, Poland) | 7 |
| Apple cv Royal Gala | Ct1670 | HE963831 AGCaV (apple, Canada) | Braeburn Ct3334 (apple, Brazil) | 6 | |
| Other | Apple | KM73720 | KY429181 (pear, Poland) | AF345892 (pear, Poland) | 6 |
| Apple | KR815876 | KF319056 (apple, Germany) | MK923754 (apple, Brazil) | 7 | |
| Apple | KF319056 | KR815875 (pear, India) | AF345892 (pear, Poland) | 7 | |
| Pear | KY176820 | KY213867 (pear, Poland) | D21829 (pear) | 6 | |
| Apple | KY176808 | JX673812 (pear, China) | JX673811 (pear, China) | 7 | |
| Pear | HM325767 | JX673812 (pear, China) | JX673811 (pear, China) | 7 | |
| Pear | AF345894 | AF345895 (pear, Poland) | AF491929 (Pear, Poland) | 7 [20] |
| Greece 1 (14) | Brazil (22) | Poland (31) | China (38) | RoW 1,2 (17) | Country |
|---|---|---|---|---|---|
| 0.221 ± 0.006 | 0.254 ± 0.007 | 0.205 ± 0.007 | 0.275 ± 0.007 | 0.244 ± 0.007 | Greece |
| - | 0.256 ± 0.007 | 0.247 ± 0.007 | 0.286 ± 0.007 | 0.257 ± 0.007 | Brazil |
| - | - | 0.185 ± 0.006 | 0.267 ± 0.007 | 0.234 ± 0.007 | Poland |
| Quince | 0.174 ± 0.007 | - | 0.290 ± 0.008 | 0.290 ± 0.008 | China |
| Botanicals | 0.271 ± 0.008 | 0.298 ± 0.008 | - | 0.260 ± 0.007 | RoW 1 |
| Pear | 0.197 ± 0.006 | 0.274 ± 0.007 | 0.207 ± 0.006 | - | |
| Apple | 0.244 ± 0.007 | 0.287 ± 0.007 | 0.245 ± 0.007 | 0.248 ± 0.008 | |
| Host | Quince 1 (5) | Botanicals (28) | Pear (35) | Apple (54) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathioudakis, M.M.; Maliogka, V.I.; Candresse, T.; Nickel, O.; Fajardo, T.V.M.; Budzyńska, D.; Hasiów-Jaroszewska, B.; Katis, N.I. Molecular Characterization of the Coat Protein Gene of Greek Apple Stem Pitting Virus Isolates: Evolution through Deletions, Insertions, and Recombination Events. Plants 2021, 10, 917. https://doi.org/10.3390/plants10050917
Mathioudakis MM, Maliogka VI, Candresse T, Nickel O, Fajardo TVM, Budzyńska D, Hasiów-Jaroszewska B, Katis NI. Molecular Characterization of the Coat Protein Gene of Greek Apple Stem Pitting Virus Isolates: Evolution through Deletions, Insertions, and Recombination Events. Plants. 2021; 10(5):917. https://doi.org/10.3390/plants10050917
Chicago/Turabian StyleMathioudakis, Matthaios M., Varvara I. Maliogka, Thierry Candresse, Osmar Nickel, Thor Vinicius Martins Fajardo, Daria Budzyńska, Beata Hasiów-Jaroszewska, and Nikolaos I. Katis. 2021. "Molecular Characterization of the Coat Protein Gene of Greek Apple Stem Pitting Virus Isolates: Evolution through Deletions, Insertions, and Recombination Events" Plants 10, no. 5: 917. https://doi.org/10.3390/plants10050917
APA StyleMathioudakis, M. M., Maliogka, V. I., Candresse, T., Nickel, O., Fajardo, T. V. M., Budzyńska, D., Hasiów-Jaroszewska, B., & Katis, N. I. (2021). Molecular Characterization of the Coat Protein Gene of Greek Apple Stem Pitting Virus Isolates: Evolution through Deletions, Insertions, and Recombination Events. Plants, 10(5), 917. https://doi.org/10.3390/plants10050917