Microbe Finder (MiFi®): Implementation of an Interactive Pathogen Detection Tool in Metagenomic Sequence Data


Abstract
:1. Introduction
2. Results
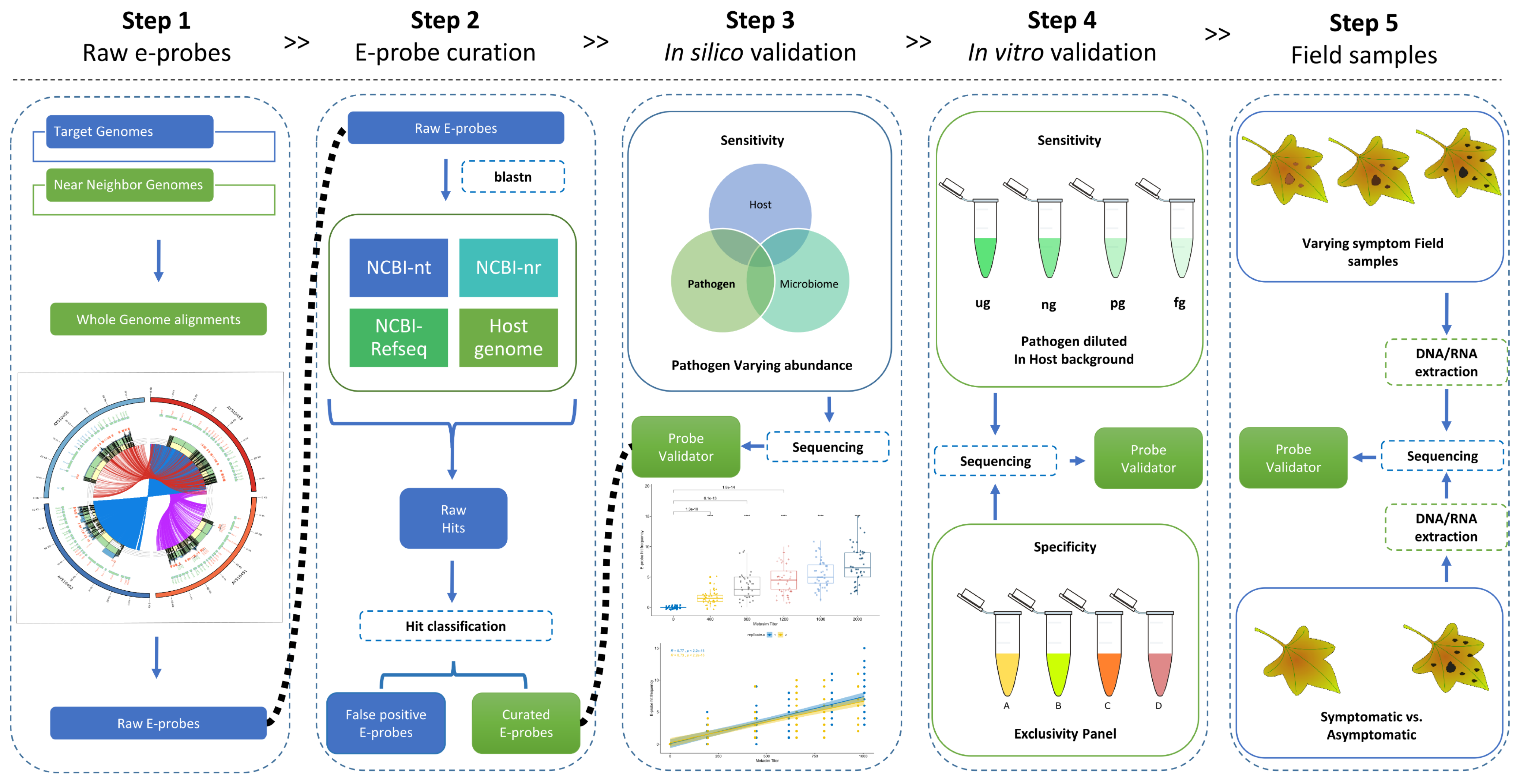
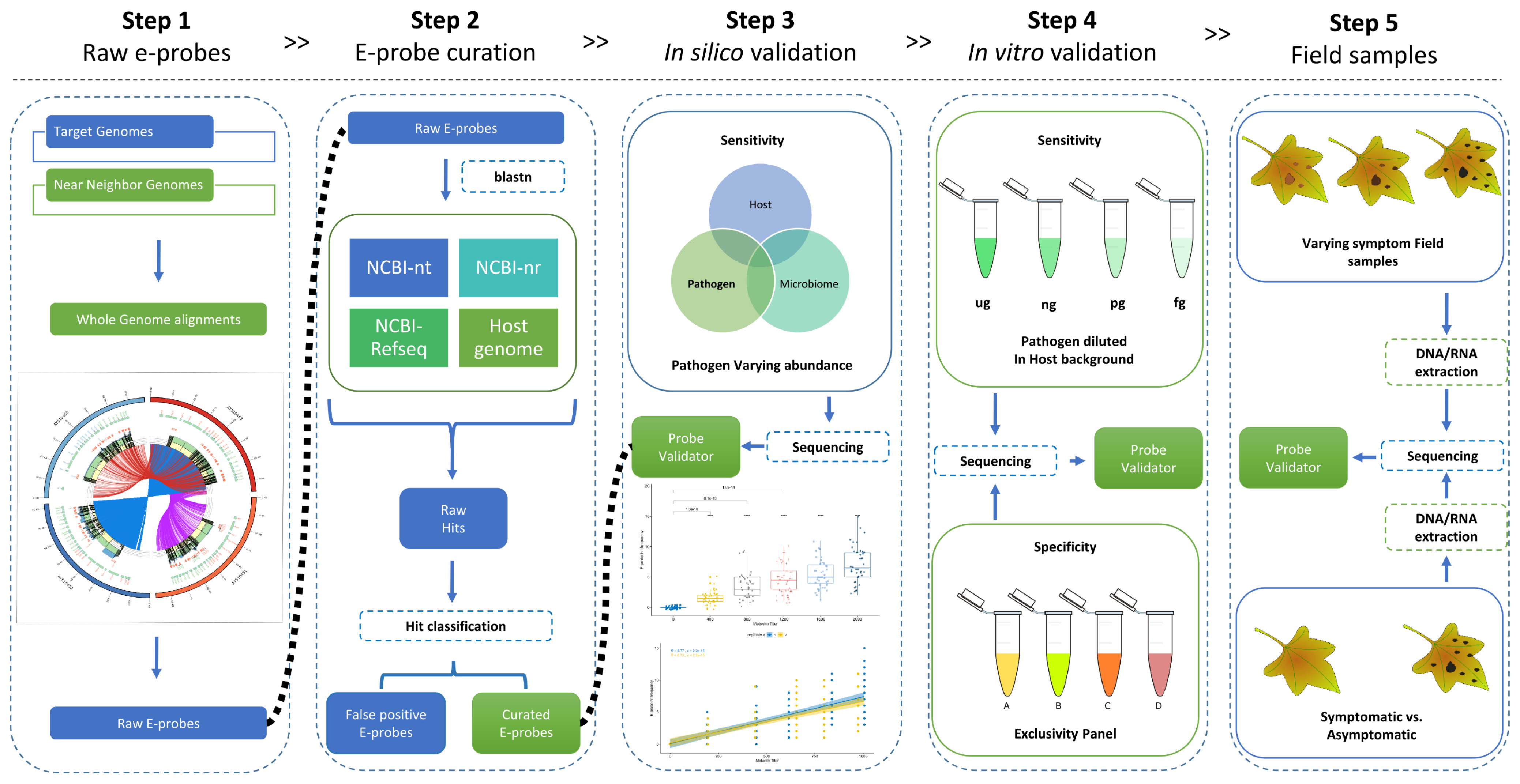
2.1. Data Selection and Raw E-Probe Design
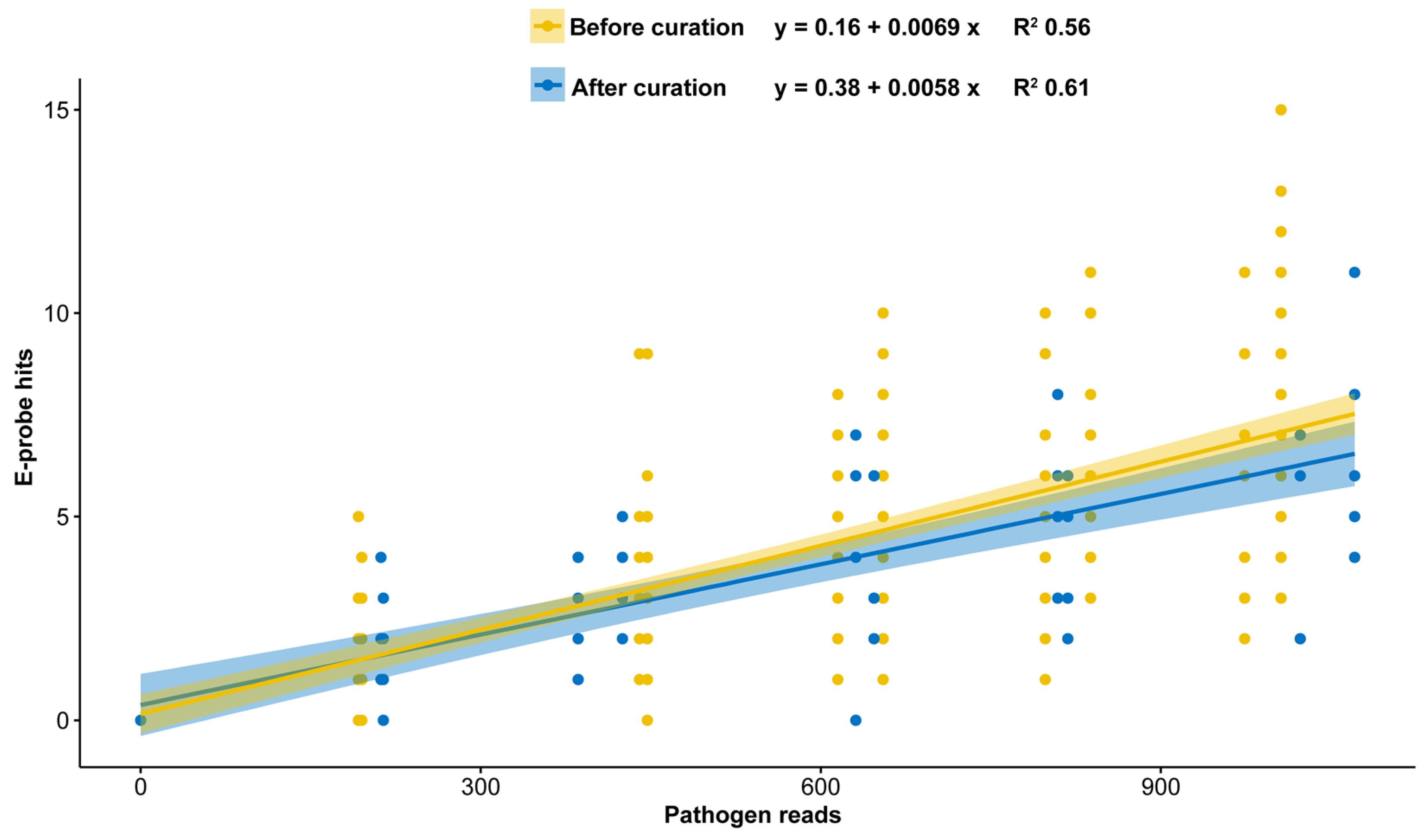
2.2. E-Probe Curation
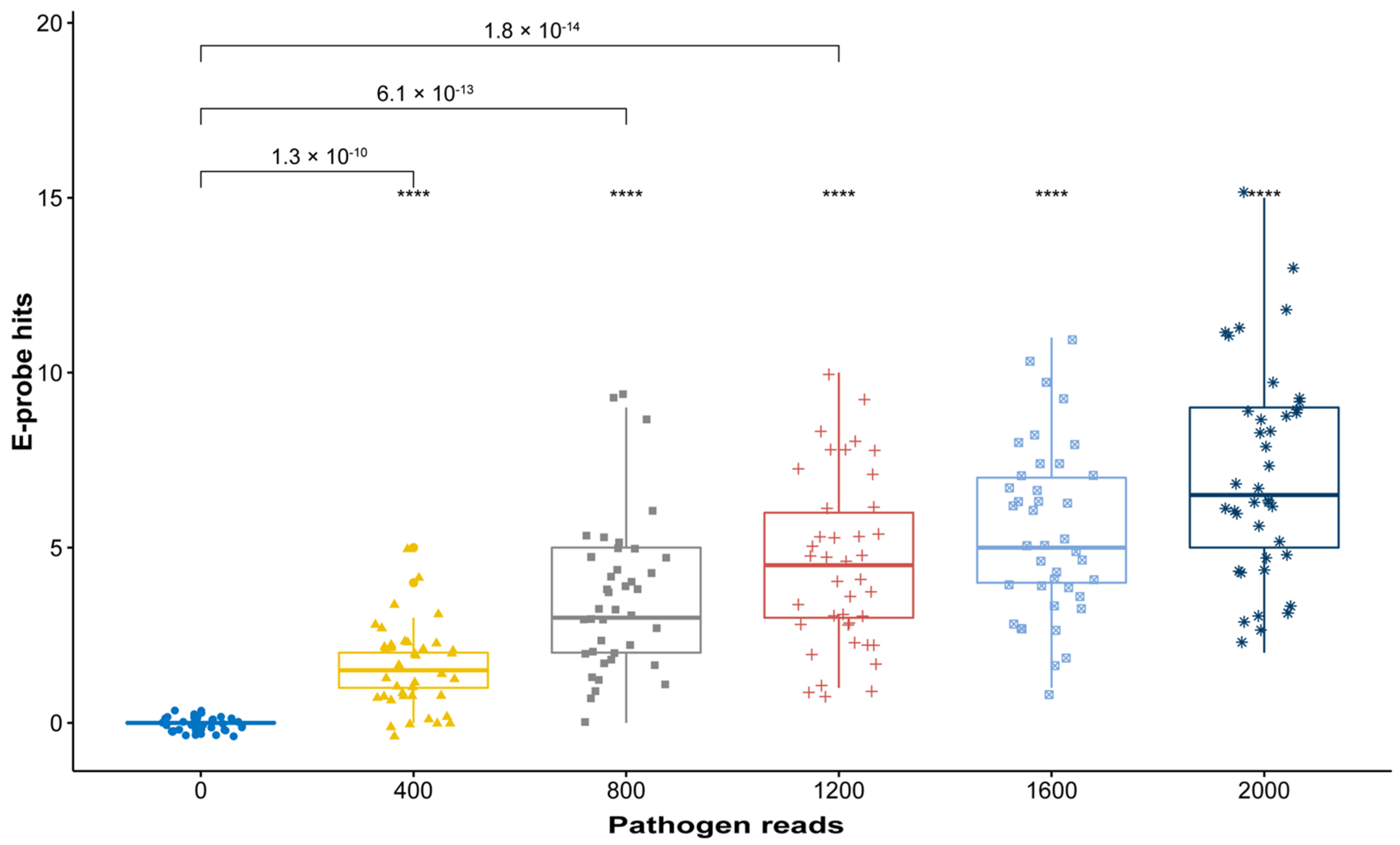
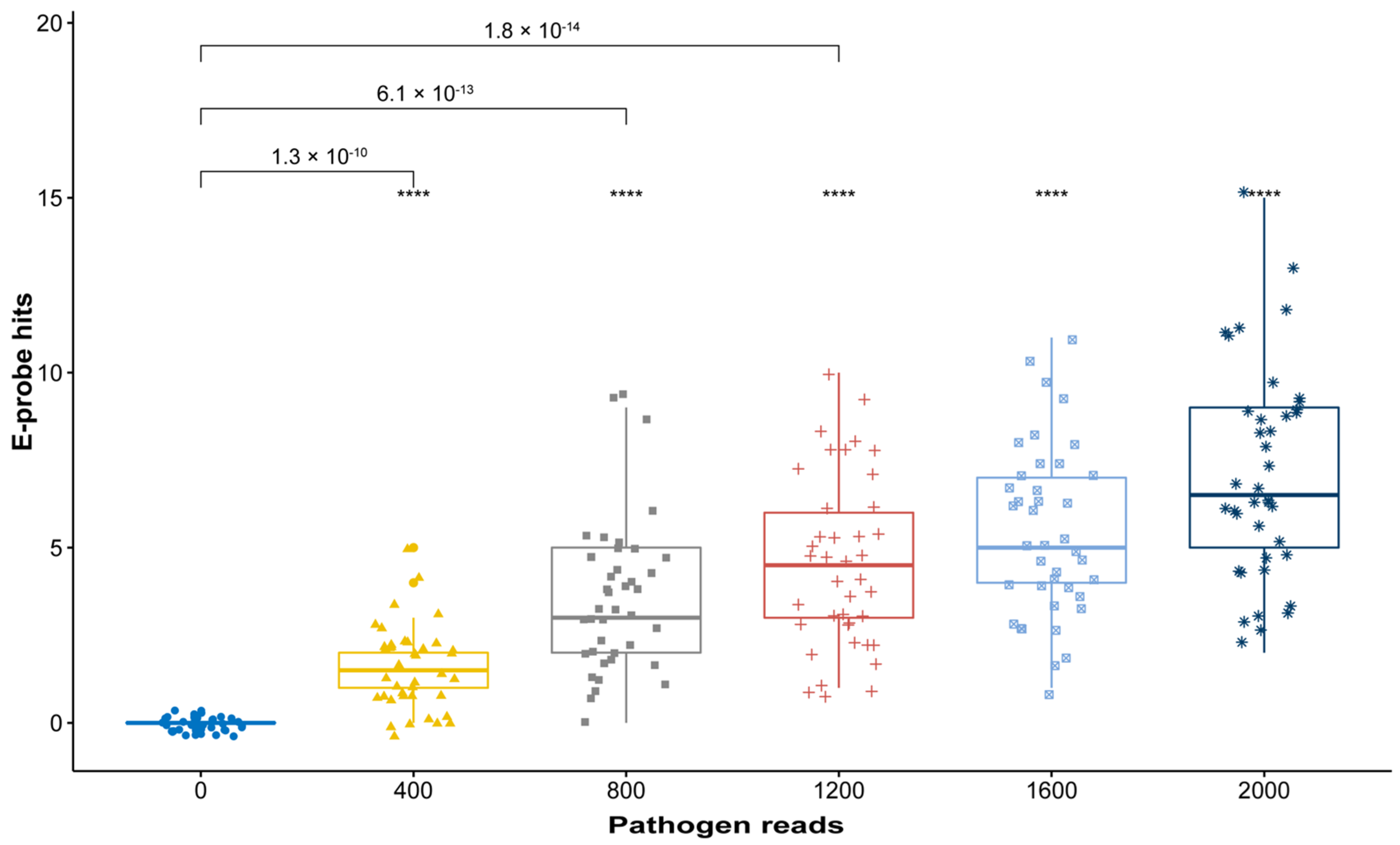
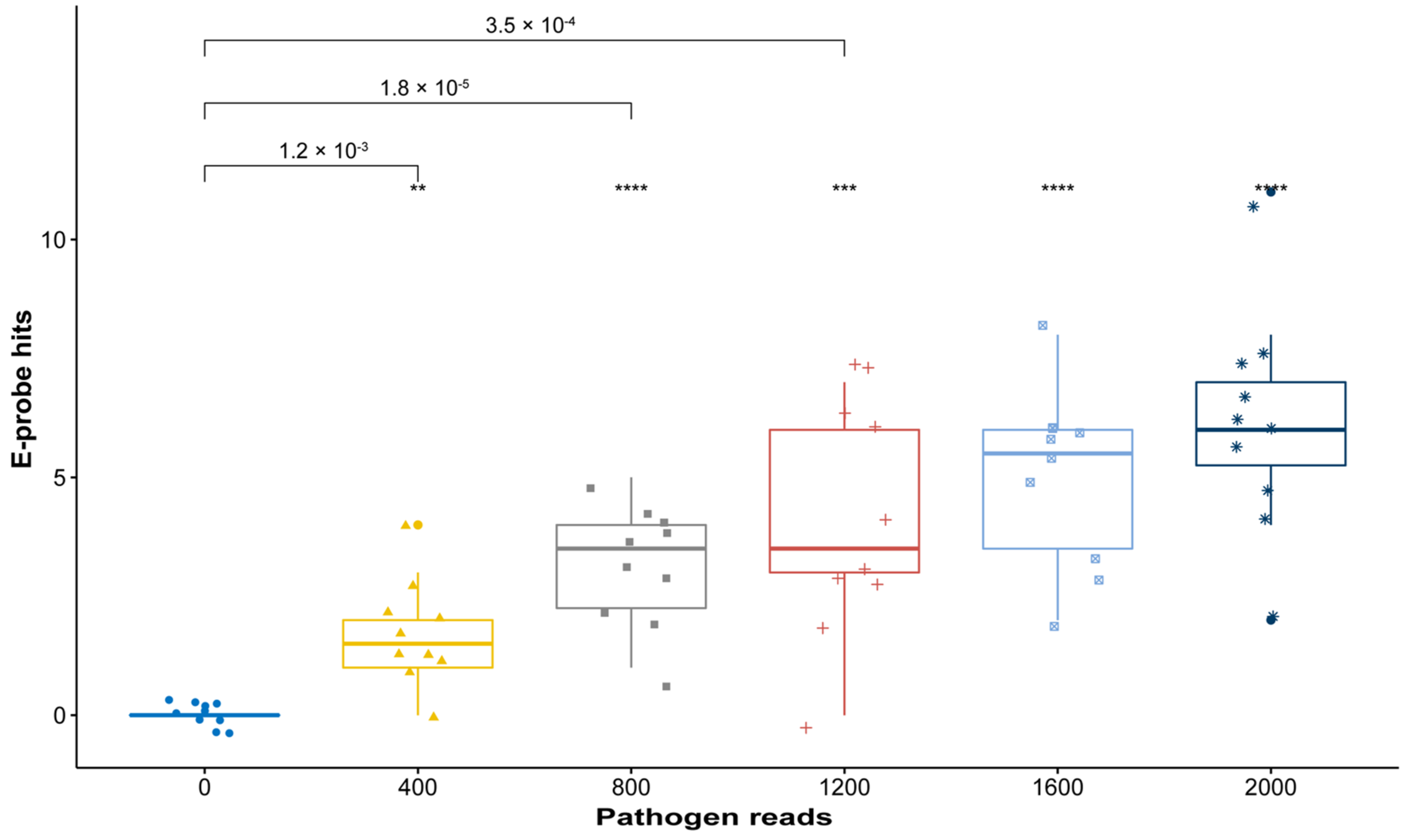
2.3. In Silico Validation with Simulated High-Throughput Sequencing (HTS) Data
2.4. In Vitro Validation: Analytical Sensitivity and Specificity
2.5. Validation with Field Sample: Diagnostic Sensitivity and Specificity
2.6. Catalogue of Pathogen E-Probes for Other Hosts
2.7. Comparison of Microbe Finder (MiFi) with Traditional Bioinformatic Tools Used for Diagnostics
3. Discussion
3.1. Data Selection and Raw E-Probe Design
3.2. E-Probe Curation
3.3. In Silico Validation
3.4. In Vitro Validation: Analytical Sensitivity (LoD) and Specificity
3.5. Validation with Field Samples: Diagnostic Sensitivity and Specificity
3.6. Comparison of MiFi with Traditional Bioinformatic Tools Used for Diagnostics
3.7. Calculations of Sequence Depth for Assured Detection of an Analyte in a Complex Metagenome
- Pathogen reads desired to detect
- Average read length (normal distribution)
- Probability
- Pathogen genome size (nts)
- Non-pathogen genome size, including host and co-habiting microbiome (nts)
4. Materials and Methods
4.1. Data Selection and Raw E-Probe Design (Step 1)
4.2. E-Probe Curation (Step 2)
4.3. In Silico Validation (Step 3)
4.4. In Vitro Validation (Step 4)
4.5. Validation with Field Samples (Step 5)
4.6. Mi Detect
4.7. Comparison of MiFi with Traditional Bioinformatic Tools Used for Diagnostics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paarlberg, P.L.; Hillberg, A.; Lee, J.G.; Mathews, K.H., Jr. Economic Impacts of Foreign Animal Disease; United States Department of Agriculture, Economic Research Service: Washington, DC, USA, 2008; Volume 57, pp. 1–8.
- Schaad, N.W.; Frederick, R.D.; Shaw, J.; Schneider, W.L.; Hickson, R.; Petrillo, M.D.; Luster, D.G. Advances in molecular-based diagnostics in meeting crop biosecurity and phytosanitary issues. Annu. Rev. Phytopathol. 2003, 41, 305–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maree, H.J.; Fox, A.; Al Rwahnih, M.; Boonham, N.; Candresse, T. Application of HTS for Routine Plant Virus Diagnostics: State of the Art and Challenges. Front. Plant Sci. 2018, 9, 1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malapi-Wight, M.; Kumar, L.; Mollov, D.S.; Foster, J. Implementation of next generation sequencing for high-throughput pathogen detection in sugarcane introductions grown in quarantine. Sugar J. 2017, 80, 22–23. [Google Scholar]
- Stobbe, A.H.; Daniels, J.; Espindola, A.S.; Verma, R.; Melcher, U.; Ochoa-Corona, F.; Garzon, C.; Fletcher, J.; Schneider, W. E-probe Diagnostic Nucleic acid Analysis (EDNA): A theoretical approach for handling of next generation sequencing data for diagnostics. J. Microbiol. Methods 2013, 94, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Espindola, A.S.; Schneider, W.; Cardwell, K.F.; Carrillo, Y.; Hoyt, P.R.; Marek, S.M.; Melouk, H.; Garzon, C.D. Inferring the presence of aflatoxin-producing Aspergillus flavus strains using RNA sequencing and electronic probes as a transcriptomic screening tool. bioRxiv 2018, 13, e0198575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espindola, A.; Cardwell, K.F. Microbe Finder (MiFi): Pathogen detection in metagenomic sequence data. In Proceedings of the Plant Health; APSNET: Saint Paul, MN, USA, 2019; Volume 109, p. S2.136. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high- throughput community sequencing data. Nature 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition - Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuniga, L.P.; Espindola, A.; Melouk, H.A.; Ali, A.; Cardwell, K.F.; Corona, F.O. Detection of cucurbit viruses in Oklahoma combining EDNA with Multiplex RT-PCR coupled with High Resolution Melting. In Proceedings of the 2017 APS Annual Meeting; APSNET: Saint Paul, MN, USA, 2017; Volume 107, p. S5.36. [Google Scholar]
- Espindola, A.; Freire-Zapata, V.; Watanabe, L.F.M.; Corona, F.O.; Cardwell, K.F. Detecting viruses and bacteria of grapevine with Microbe Finder (MiFi) and an Oxford Nanopore sequencer. In Proceedings of the Plant Health; APSNET: Saint Paul, MN, USA, 2019; p. S2.138. [Google Scholar]
- Espindola, A. Massively Parallel Sequencing (Mps) As a Diagnostic and Forensic Analysis Tool for Important Fungi and Chromista Plant Pathogens. Ph.D. Thesis, Oklahoma State University, Stillwater, OK, USA, 2013. [Google Scholar]
- Espindola, A.; Roy, A.; Mavrodieva, V.A.; Cardwell, K.F. E-probe: A new diagnostic tool for detection of Dichorhaviruses associated with Citrus leprosis syndrome. In Proceedings of the Plant Health; APSNET: Saint Paul, MN, USA, 2019; Volume 109, p. S2.138. [Google Scholar]
- Bocsanczy, A.M.; Espindola, A.; Norman, D.J.; Cardwell, K.F. E-probes development for rapid, sensitive and specific pathogen detection in blueberries. In Proceedings of the ICPP Boston; APSNET: Saint Paul, MN, USA, 2018; Volume 108, p. S1.301. [Google Scholar]
- Proano, M.F.; Espindola, A.; Garzon, C.D. Detection of multiple oomycetes in metagenomic data by Using E-probe Detection of Nucleic Analysis (EDNA). In Proceedings of the ICPP Boston; APSNET: Saint Paul, MN, USA, 2018; Volume 108, p. S1.286. [Google Scholar]
- Cardwell, K.; Dennis, G.; Flannery, A.R.; Fletcher, J.; Luster, D.; Nakhla, M.; Rice, A.; Shiel, P.; Stack, J.; Walsh, C.; et al. Principles of Diagnostic Assay Validation for Plant Pathogens: A Basic Review of Concepts. Plant Health Prog. 2018, 19, 272–278. [Google Scholar] [CrossRef]
- Fegan, M.; Taghavi, M.; Sly, L.I.; Hayward, A.C. Phylogeny, Diversity and Molecular Diagnostics of Ralstonia solanacearum. In Bacterial Wilt Disease: Molecular and Ecological Aspects; Prior, P., Allen, C., Elphinstone, J., Eds.; Springer: Berlin/Heidelberg, Germany, 1998; pp. 19–33. ISBN 9783662035924. [Google Scholar]
- Espindola, A.; Schneider, W.; Hoyt, P.R.; Marek, S.M.; Garzon, C. A new approach for detecting fungal and oomycete plant pathogens in next generation sequencing metagenome data utilising electronic probes. Int. J. Data Min. Bioinform. 2015, 12, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Espindola, A.; Habiger, J.; Cardwell, K. Metagenome sequence calculator for effective pathogen detection. In Proceedings of the Plant Health; APSNET: Saint Paul, MN, USA, 2019; Volume 109, p. S2.138. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Chu, J.; Warren, R.L.; Birol, I. NanoSim: Nanopore sequence read simulator based on statistical characterization. Gigascience 2017, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Sczyrba, A.; Hofmann, P.; Belmann, P.; Koslicki, D.; Janssen, S.; Dröge, J.; Gregor, I.; Majda, S.; Fiedler, J.; Dahms, E.; et al. Critical Assessment of Metagenome Interpretation-a benchmark of metagenomics software. Nat. Methods 2017, 14, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species Name | NCBI Taxon ID | E-Probe Sequences | MRAD (LoD) |
|---|---|---|---|
| Grapevine Fanleaf Virus GFD | 12274 | 6 | 0.1272% |
| Grapevine Virus A RWC | 35288 | 5 | 0.0512% |
| Grapevine Virus B RWC | 35289 | 11 | 0.005% |
| Grapevine Leafroll-associated virus 1 GLD | 47985 | 16 | 0.0095% |
| Grapevine Leafroll-associated virus 2 GLD | 64003 | 10 | 0.0174% |
| Grapevine Leafroll-associated Virus 3 GLD | 55951 | 5 | 0.0102% |
| Grapevine Leafroll-associated Virus 4(4) GLD | 70177 | 9 | 0.0089% |
| Grapevine Leafroll-associated Virus 4(5) GLD | 71032 | 3 | 0.2521% |
| Grapevine Leafroll-associated Virus 4(6) GLD | 203168 | 2 | 0.3971% |
| Grapevine Leafroll-associated Virus 7 GLD | 217615 | 8 | 0.01% |
| Grapevine Leafroll-associated Virus 4(9) GLD | 184610 | 7 | 0.0105% |
| Grapevine Leafroll-associated Virus 4(Pr) GLD | 367121 | 9 | 0.01% |
| Grapevine Leafroll-associated Virus 4(Car) GLD | 659661 | 8 | 0.0094% |
| Grapevine Leafroll-associated Virus 13 GLD | 1815581 | 22 | 0.0017% |
| Arabis Mosaic Virus MD | 12271 | 7 | 0.1309% |
| Tomato Ringspot Virus YV | 12280 | 7 | 0.04% |
| Tobacco Ringspot Virus TRD | 12282 | 8 | 0.1304% |
| Grapevine red blotch-associated virus RBD | 1381007 | 2 | 0.4087% |
| Xylella fastidiosaPD | 644356 | 4034 | 0.0022% |
| Agrobacterium vitisCG | 373 | 14,236 | 0.0011% |
| Candidatus Phytoplasma solani | 69896 | 83 | 0.0362% |
| Candidatus Phytoplasma australiense | 59748 | 78 | 0.0257% |
| Candidatus Phytoplasma aurantifolia | 180978 | 122 | 0.027% |
| Type | Host | Pathogens | Taxonomic Level |
|---|---|---|---|
| Plant | Grapevine | 31 | species, strain |
| Citrus | 43 | Species, strain | |
| Rose | 22 | species | |
| Cucurbits | 15 | species | |
| Wheat | 23 | species | |
| Rhododendron | 1 | species | |
| Blueberry | 3 | strain | |
| Animal | Swine gut microbiome | 35 | family |
| Bovine respiratory disease complex | 5 | species | |
| Human Pathogens on Plants | Food-borne pathogens | 5 | species |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espindola, A.S.; Cardwell, K.F. Microbe Finder (MiFi®): Implementation of an Interactive Pathogen Detection Tool in Metagenomic Sequence Data. Plants 2021, 10, 250. https://doi.org/10.3390/plants10020250
Espindola AS, Cardwell KF. Microbe Finder (MiFi®): Implementation of an Interactive Pathogen Detection Tool in Metagenomic Sequence Data. Plants. 2021; 10(2):250. https://doi.org/10.3390/plants10020250
Chicago/Turabian StyleEspindola, Andres S., and Kitty F. Cardwell. 2021. "Microbe Finder (MiFi®): Implementation of an Interactive Pathogen Detection Tool in Metagenomic Sequence Data" Plants 10, no. 2: 250. https://doi.org/10.3390/plants10020250
APA StyleEspindola, A. S., & Cardwell, K. F. (2021). Microbe Finder (MiFi®): Implementation of an Interactive Pathogen Detection Tool in Metagenomic Sequence Data. Plants, 10(2), 250. https://doi.org/10.3390/plants10020250