
Development of SSR Databases Available for Both NGS and Capillary Electrophoresis in Apple, Pear and Tea

 , ,
, ,
Abstract
1. Introduction
2. Results
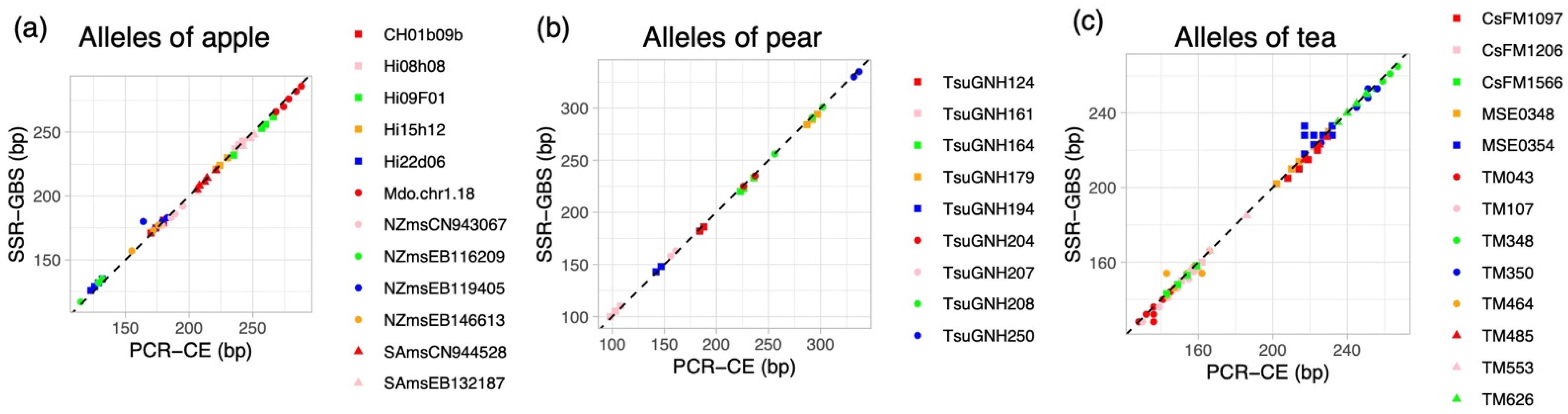
2.1. Genotyping by PCR-CE
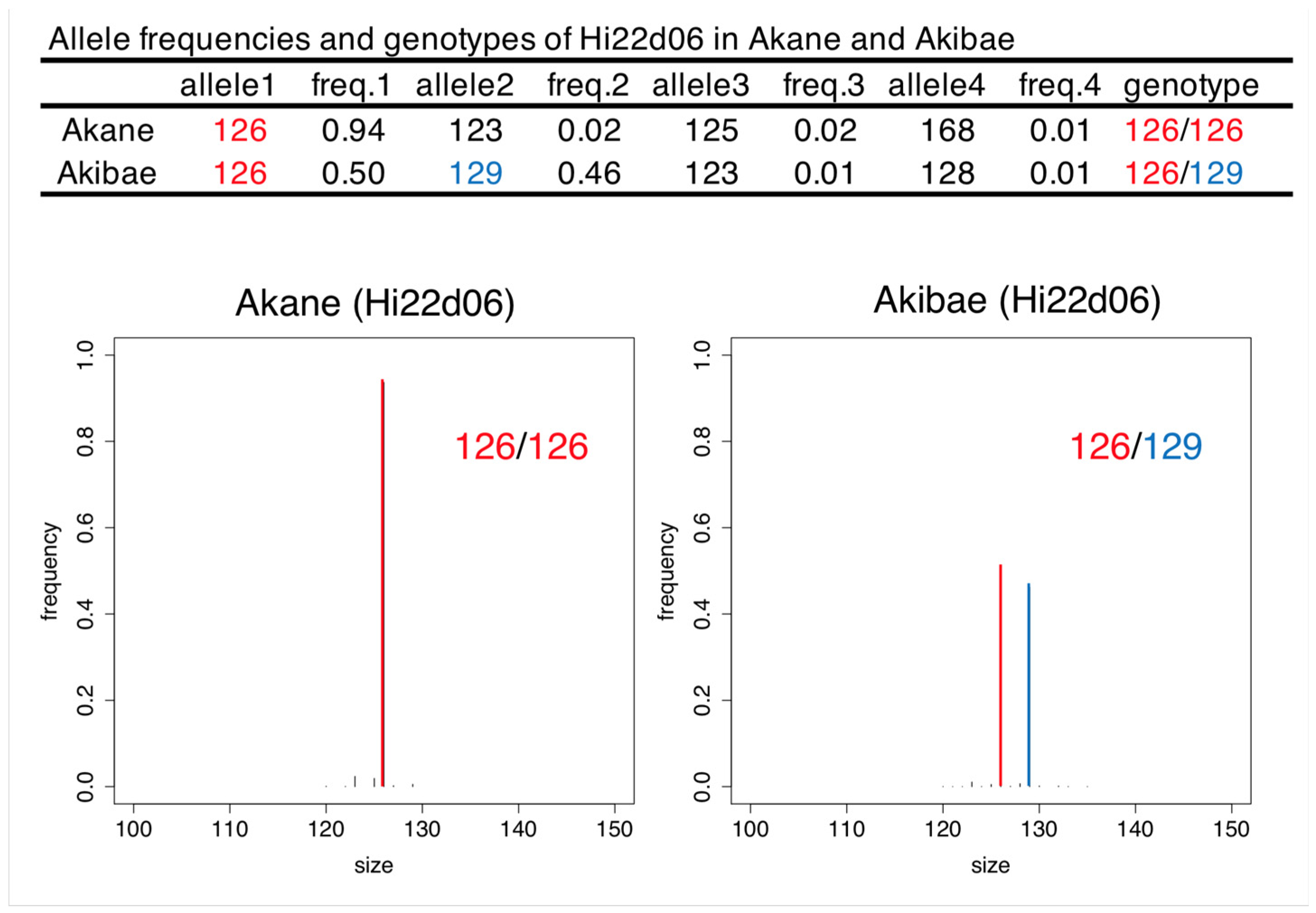
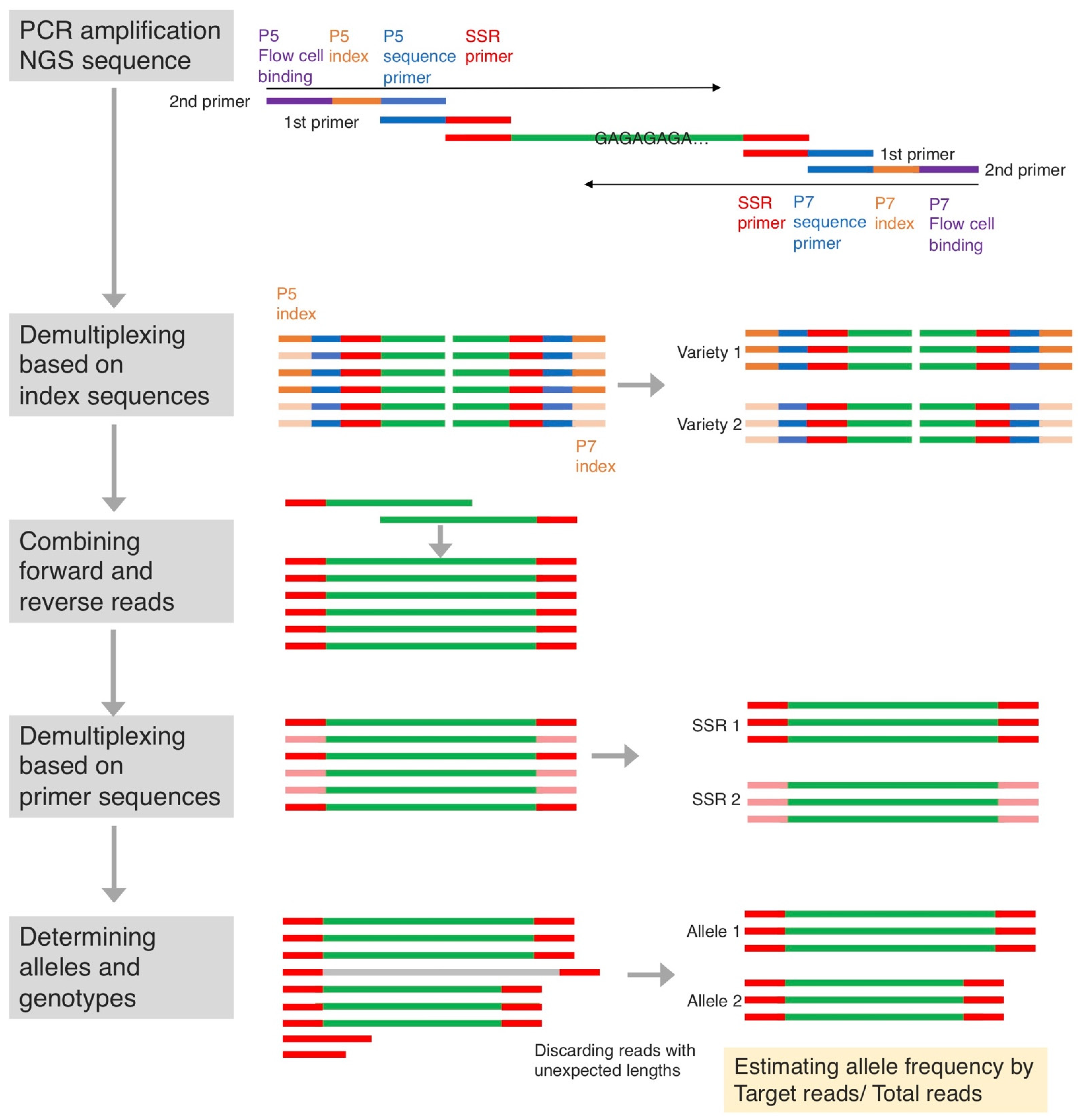
2.2. Genotyping by SSR-GBS
2.3. Automated Genotyping in SSR-GBS and Repeatability
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. SSR Genotyping for PCR-CE
4.3. SSR Genotyping Using SSR-GBS
4.4. Automated Genotyping in SSR-GBS and Repeatability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luby, J.J.; Shaw, D.V. Does marker-assisted selection make dollars and sense in a fruit breeding program? HortScience 2001, 36, 872–879. [Google Scholar] [CrossRef]
- Janick, J. History of Fruit Breeding. Fruit Veg. Ceral Sci. Biotechnol. 2011, 5, 1–7. [Google Scholar]
- Saito, T. Advances in Japanese pear breeding in Japan. Breed. Sci. 2016, 66, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Sato, A. Advances in table grape breeding in Japan. Breed. Sci. 2016, 66, 34–45. [Google Scholar] [CrossRef]
- Endo, T.; Fujii, H.; Yoshioka, T.; Omura, M.; Shimada, T. TaqMan-MGB SNP genotyping assay to identify 48 citrus cultivars distributed in the Japanese market. Breed. Sci. 2020, 70, 363–372. [Google Scholar] [CrossRef]
- Scribner, K.T.; Pearce, J.M. Microsatellites: Evolutionary and methodological background and empirical applications at individual, population, and phylogenetic levels. In Molecular Methods in Ecology; Blackwell Science Limited: London, UK, 2000; pp. 235–273. [Google Scholar]
- Sunnucks, P. Efficient genetic markers for population biology. Trends Ecol. Evol. 2000, 15, 199–203. [Google Scholar] [CrossRef]
- Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, K.A.; Toonen, R.J. Microsatellites for ecologists: A practical guide to using and evaluating microsatellite markers. Ecol. Lett. 2006, 9, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Clayton, T.M.; Whitaker, J.P.; Sparkes, R.; Gill, P. Analysis and interpretation of mixed forensic stains using DNA STR profiling. Forensic Sci. Int. 1998, 91, 55–70. [Google Scholar] [CrossRef]
- Cipriani, G.; Spadotto, A.; Jurman, I.; Di Gaspero, G.; Crespan, M.; Meneghetti, S.; Frare, E.; Vignani, R.; Cresti, M.; Morgante, M. The SSR-based molecular profile of 1005 grapevine (Vitis vinifera L.) accessions uncovers new synonymy and parentages, and reveals a large admixture amongst varieties of different geographic origin. Theor. Appl. Genet. 2010, 121, 1569–1585. [Google Scholar] [CrossRef]
- Moriya, S.; Iwanami, H.; Okada, K.; Yamamoto, T.; Abe, K. A practical method for apple cultivar identification and parent-offspring analysis using simple sequence repeat markers. Euphytica 2011, 177, 135–150. [Google Scholar] [CrossRef]
- Sawamura, Y.; Takada, N.; Yamamoto, T.; Saito, T.; Kimura, T.; Kotobuki, K. Identification of parent-offspring relationships in 55 Japanese pear cultivars using S-RNase allele and SSR markers. J. Jpn. Soc. Hortic. Sci. 2008, 77, 364–373. [Google Scholar] [CrossRef][Green Version]
- Kimura, T.; Shi, Y.Z.; Shoda, M.; Kotobuki, K.; Matsuta, N.; Hayashi, T.; Ban, Y.; Yamamoto, T. Identification of Asian pear varieties by SSR analysis. Breed. Sci. 2002, 52, 115–121. [Google Scholar] [CrossRef]
- Leigh, F.; Lea, V.; Law, J.; Wolters, P.; Powell, W.; Donini, P. Assessment of EST-and genomic microsatellite markers for variety discrimination and genetic diversity studies in wheat. Euphytica 2003, 133, 359–366. [Google Scholar] [CrossRef]
- Hosseinzadeh-Colagar, A.; Haghighatnia, M.J.; Amiri, Z.; Mohadjerani, M.; Tafrihi, M. Microsatellite (SSR) amplification by PCR usually led to polymorphic bands: Evidence which shows replication slippage occurs in extend or nascent DNA strands. Mol. Biol. Res. Commun. 2016, 5, 167. [Google Scholar]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, K.; Hirakawa, H.; Isobe, S. Analytical workflow of double-digest restriction site-associated DNA sequencing based on empirical and in silico optimization in tomato. DNA Res. 2016, 23, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Du, F.K.; Xu, F.; Qu, H.; Feng, S.; Tang, J.; Wu, R. Exploiting the transcriptome of Euphrates Poplar, Populus euphratica (Salicaceae) to develop and characterize new EST-SSR markers and construct an EST-SSR database. PLoS ONE 2013, 8, e61337. [Google Scholar] [CrossRef]
- Dubey, H.; Rawal, H.C.; Rohilla, M.; Lama, U.; Kumar, P.M.; Bandyopadhyay, T.; Gogoi, M.; Singh, N.K.; Mondal, T.K. TeaMiD: A comprehensive database of simple sequence repeat markers of tea. Database 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.; Takada, N.; Terakami, S.; Kato, H.; Inoue, H.; Takeuchi, Y.; Saito, T. Estimation of effective pollen dispersal distance for cross-pollination in chestnut orchards by microsatellite-based paternity analyses. Sci. Hortic. (Amsterdam) 2019, 250, 89–93. [Google Scholar] [CrossRef]
- Silfverberg-Dilworth, E.; Matasci, C.L.; Van de Weg, W.E.; Van Kaauwen, M.P.W.; Walser, M.; Kodde, L.P.; Soglio, V.; Gianfranceschi, L.; Durel, C.E.; Costa, F. Microsatellite markers spanning the apple (Malus x domestica Borkh.) genome. Tree Genet. Genomes 2006, 2, 202–224. [Google Scholar] [CrossRef]
- Matsuki, Y.; Isagi, Y.; Suyama, Y. The determination of multiple microsatellite genotypes and DNA sequences from a single pollen grain. Mol. Ecol. Notes 2007, 7, 194–198. [Google Scholar] [CrossRef]
- Suyama, Y. Procedure for single-pollen genotyping. In Single-Pollen Genotyping; Springer: Berlin/Heidelberg, Germany, 2011; pp. 7–15. [Google Scholar]
- Tibihika, P.D.; Curto, M.; Dornstauder-Schrammel, E.; Winter, S.; Alemayehu, E.; Waidbacher, H.; Meimberg, H. Application of microsatellite genotyping by sequencing (SSR-GBS) to measure genetic diversity of the East African Oreochromis niloticus. Conserv. Genet. 2019, 20, 357–372. [Google Scholar] [CrossRef]
- Šarhanová, P.; Pfanzelt, S.; Brandt, R.; Himmelbach, A.; Blattner, F.R. SSR-seq: Genotyping of microsatellites using next-generation sequencing reveals higher level of polymorphism as compared to traditional fragment size scoring. Ecol. Evol. 2018, 8, 10817–10833. [Google Scholar] [CrossRef]
- Curto, M.; Winter, S.; Seiter, A.; Schmid, L.; Scheicher, K.; Barthel, L.M.F.; Plass, J.; Meimberg, H. Application of a SSR-GBS marker system on investigation of European Hedgehog species and their hybrid zone dynamics. Ecol. Evol. 2019, 9, 2814–2832. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Paterson, I.G.; Fraser, B.A.; Watson, B.; Bradbury, I.R.; Nadukkalam Ravindran, P.; Reznick, D.; Beiko, R.G.; Bentzen, P. MEGASAT: Automated inference of microsatellite genotypes from sequence data. Mol. Ecol. Resour. 2017, 17, 247–256. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Daccord, N.; Celton, J.M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; Van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef]
- Shirasawa, K.; Itai, A.; Isobe, S. Chromosome-scale genome assembly of Japanese pear (Pyrus pyrifolia) variety ‘Nijisseiki’. DNA Res. 2021, 28. [Google Scholar] [CrossRef]
- Wei, C.; Yang, H.; Wang, S.; Zhao, J.; Liu, C.; Gao, L.; Xia, E.; Lu, Y.; Tai, Y.; She, G. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4151–E4158. [Google Scholar] [CrossRef] [PubMed]
- Darby, B.J.; Erickson, S.F.; Hervey, S.D.; Ellis-Felege, S.N. Digital fragment analysis of short tandem repeats by high-throughput amplicon sequencing. Ecol. Evol. 2016, 6, 4502–4512. [Google Scholar] [CrossRef] [PubMed]
- Neophytou, C.; Torutaeva, E.; Winter, S.; Meimberg, H.; Hasenauer, H.; Curto, M. Analysis of microsatellite loci in tree of heaven (Ailanthus altissima (Mill.) Swingle) using SSR-GBS. Tree Genet. Genomes 2018, 14, 82. [Google Scholar] [CrossRef]
- Lepais, O.; Chancerel, E.; Boury, C.; Salin, F.; Manicki, A.; Taillebois, L.; Dutech, C.; Aissi, A.; Bacles, C.F.E.; Daverat, F. Fast sequence-based microsatellite genotyping development workflow. PeerJ 2020, 8, e9085. [Google Scholar] [CrossRef]
- Bornman, D.M.; Hester, M.E.; Schuetter, J.M.; Kasoji, M.D.; Minard-Smith, A.; Barden, C.A.; Nelson, S.C.; Godbold, G.D.; Baker, C.H.; Yang, B.; et al. Short-read, high-throughput sequencing technology for STR genotyping. Biotech. Rapid Dispatches 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef] [PubMed]
- Celton, J.-M.; Tustin, D.S.; Chagné, D.; Gardiner, S.E. Construction of a dense genetic linkage map for apple rootstocks using SSRs developed from Malus ESTs and Pyrus genomic sequences. Tree Genet. Genomes 2009, 5, 93–107. [Google Scholar] [CrossRef]
- Kunihisa, M.; Moriya, S.; Abe, K.; Okada, K.; Haji, T.; Hayashi, T.; Kim, H.; Nishitani, C.; Terakami, S.; Yamamoto, T. Identification of QTLs for fruit quality traits in Japanese apples: QTLs for early ripening are tightly related to preharvest fruit drop. Breed. Sci. 2014, 64, 240–251. [Google Scholar] [CrossRef]
- Liebhard, R.; Gianfranceschi, L.; Koller, B.; Ryder, C.D.; Tarchini, R.; Van de Weg, E.; Gessler, C. Development and characterisation of 140 new microsatellites in apple (Malus x domestica Borkh.). Mol. Breed. 2002, 10, 217–241. [Google Scholar] [CrossRef]
- Van Dyk, M.M.; Soeker, M.K.; Labuschagne, I.F.; Rees, D.J.G. Identification of a major QTL for time of initial vegetative budbreak in apple (Malus x domestica Borkh.). Tree Genet. Genomes 2010, 6, 489–502. [Google Scholar] [CrossRef]
- Yamamoto, T.; Terakami, S.; Hosaka, F.; Fujii, H.; Kunihisa, M.; Takada, N.; Saito, T.; Nishitani, C. DNA profiling of Japanese pear cultivars by SSR markers with tetra- and penta-nucleotide motifs. DNA Polymorph. 2021, 20, 58–61. (In Japanese) [Google Scholar]
- Ma, J.; Zhou, Y.; Ma, C.; Yao, M.; Jin, J.; Wang, X.; Chen, L. Identification and characterization of 74 novel polymorphic EST-SSR markers in the tea plant, Camellia sinensis (Theaceae). Am. J. Bot. 2010, 97, e153–e156. [Google Scholar] [CrossRef]
- Tan, L.Q.; Wang, L.Y.; Wei, K.; Zhang, C.C.; Wu, L.Y.; Qi, G.N.; Cheng, H.; Zhang, Q.; Cui, Q.M.; Liang, J.B. Floral transcriptome sequencing for SSR marker development and linkage map construction in the tea plant (Camellia sinensis). PLoS ONE 2013, 8, e81611. [Google Scholar]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Apple | Pear | Tea | ||||||
|---|---|---|---|---|---|---|---|---|
| ID | Variety | Accession no. a | ID | Variety | Accession no. a | ID | Variety | Accession no. a |
| APPLE01 | Akane | JP169082 | PEAR01 | Akiakari | JP118536 | TEA01 | Ryofu | NA |
| APPLE02 | Akibae | NA | PEAR02 | Akizuki | JP118538 | TEA02 | Cha Chukanbohon No 3 | NA |
| APPLE03 | Iwakami | JP114453 | PEAR03 | Atago | JP113570 | TEA03 | Harumidori | NA |
| APPLE04 | Indo | JP169681 | PEAR04 | Amanogawa | JP113562 | TEA04 | Sofu | NA |
| APPLE05 | Orin | JP172644 | PEAR05 | Oushuu | JP118539 | TEA05 | Cha Chukanbohon No 4 | JP232165 |
| APPLE06 | Ozenokurenai | NA | PEAR06 | Okusankichi | JP113634 | TEA06 | Cha Chukanbohon No 5 | JP232219 |
| APPLE07 | Gala | JP114085 | PEAR07 | Natsushizuku | JP230439 | TEA07 | Cha Chukanbohon No 6 | NA |
| APPLE08 | Kio | NA | PEAR08 | Kumoi | JP113623 | TEA08 | Sun Rouge | NA |
| APPLE09 | Kizashi | JP114525 | PEAR09 | Kousui | JP113619 | TEA09 | Shuntaro | NA |
| APPLE10 | Kitakami | JP114148 | PEAR10 | Shuugyoku | JP113707 | TEA10 | Saeakari | NA |
| APPLE11 | Kitaro | NA | PEAR11 | Shuurei | JP118537 | TEA11 | Nanmei | NA |
| APPLE12 | Kinsei | JP172634 | PEAR12 | Shinkou | JP113657 | TEA12 | Seimei | NA |
| APPLE13 | Jonathan | JP169679 | PEAR13 | Shinsui | JP113660 | TEA13 | Sayamakaori | NA |
| APPLE14 | Kotaro | NA | PEAR14 | Shinsei | JP113694 | TEA14 | Yabukita | JP168695 |
| APPLE15 | Golden Delicious | JP116722 | PEAR15 | Suisei | JP113665 | TEA15 | Asatsuyu | NA |
| APPLE16 | Sansa | JP114526 | PEAR16 | Chikusui | JP113716 | TEA16 | Okumidori | NA |
| APPLE17 | Santaro | NA | PEAR17 | Choujuurou | JP113574 | TEA17 | Kanayamidori | NA |
| APPLE18 | Shinano Sweet | NA | PEAR18 | Niitaka | JP113630 | TEA18 | Saemidori | JP171014 |
| APPLE19 | Jonagold | JP172630 | PEAR19 | Nijisseiki | JP113631 | TEA19 | Yutakamidori | JP168749 |
| APPLE20 | Sensyu | JP114288 | PEAR20 | Hakkou | JP113585 | TEA20 | Kiyoka | NA |
| APPLE21 | Chinatsu | NA | PEAR21 | Hayatama | JP113591 | TEA21 | MK5601 | NA |
| APPLE22 | Tsugaru | JP172651 | PEAR22 | Hougetsu | JP113720 | TEA22 | Kanaemaru | NA |
| APPLE23 | Delicious | JP114041 | PEAR23 | Housui | JP113598 | TEA23 | Yachaken01 | NA |
| APPLE24 | Hatsuaki | JP169705 | PEAR24 | Yasato | JP113718 | TEA24 | Yachaken02 | NA |
| APPLE25 | Himekami | JP169714 | PEAR25 | Hatsumaru | NA | TEA25 | Yachaken09 | NA |
| APPLE26 | Fuji | JP114078 | PEAR26 | Rinka | NA | TEA26 | Yachaken10 | NA |
| APPLE27 | Hokuto | JP169715 | PEAR27 | Hoshiakari | NA | TEA27 | Kokken01 | NA |
| APPLE28 | Maypole | NA | PEAR28 | Narumi | NA | TEA28 | Danshin37 | NA |
| APPLE29 | Mori-no-kagayaki | NA | PEAR29 | Kanta | NA | TEA29 | Miyazaki39 | NA |
| APPLE30 | Yoko | JP114416 | TEA30 | Miyazaki40 | NA | |||
| APPLE31 | Ruby Sweet | NA | TEA31 | Okuharuka | NA | |||
| APPLE32 | Rose Pearl | NA | TEA32 | Kirari31 | NA | |||
| APPLE33 | JM1 | NA | TEA33 | Sainomidori | NA | |||
| APPLE34 | JM2 | NA | TEA34 | Sakimidori | NA | |||
| APPLE35 | JM5 | NA | TEA35 | Nagomiyutaka | NA | |||
| APPLE36 | JM7 | NA | TEA36 | Haruto34 | NA | |||
| APPLE37 | JM8 | NA | TEA37 | Harunonagori | NA | |||
| APPLE38 | Morioka 66 | NA | TEA38 | Harumoegi | NA | |||
| APPLE39 | Beniminori | NA | TEA39 | Miyamakaori | NA | |||
| APPLE40 | Morioka 68 | NA | TEA40 | Musashikaori | NA | |||
| APPLE41 | Morioka 69 | NA | TEA41 | Yumekaori | NA | |||
| APPLE42 | Kinshu | NA | TEA42 | Yumewakaba | NA | |||
| APPLE43 | Morioka 71 | NA | TEA43 | Benifuki | NA | |||
| TEA44 | Sayamaakari | NA | ||||||
| Marker | Plant | No. of Alleles | HO | HE | PIC |
|---|---|---|---|---|---|
| CH01b09b a | Apple | 3 | 0.59 | 0.67 | 0.52 |
| Hi08h08 | Apple | 3 | 0.56 | 0.54 | 0.47 |
| Hi09f01 | Apple | 4 | 0.79 | 0.63 | 0.57 |
| Hi15h12 | Apple | 3 | 0.26 | 0.29 | 0.26 |
| Hi22d06 | Apple | 5 | 0.79 | 0.74 | 0.69 |
| Mdo.chr1.18 | Apple | 5 | 0.63 | 0.64 | 0.57 |
| NZmsCN943067 | Apple | 5 | 0.61 | 0.51 | 0.46 |
| NZmsEB116209 | Apple | 3 | 0.70 | 0.66 | 0.58 |
| NzmsEB119405 a | Apple | 4 | 0.52 | 0.61 | 0.48 |
| NzmsEB146613 | Apple | 3 | 0.74 | 0.63 | 0.56 |
| SamsCN944528 | Apple | 5 | 0.65 | 0.61 | 0.55 |
| SamsEB132187 | Apple | 3 | 0.65 | 0.60 | 0.52 |
| Averages for apple markers | 3.8 | 0.62 | 0.59 | 0.52 | |
| TsuGNH124 | Pear | 2 | 0.55 | 0.49 | 0.37 |
| TsuGNH161 | Pear | 3 | 0.62 | 0.60 | 0.52 |
| TsuGNH164 a | Pear | 3 | 0.57 | 0.41 | 0.49 |
| TsuGNH179 | Pear | 3 | 0.83 | 0.59 | 0.52 |
| TsuGNH184 a | Pear | 2 | 0.50 | 0.48 | 0.37 |
| TsuGNH194 | Pear | 2 | 0.35 | 0.37 | 0.30 |
| TsuGNH204 | Pear | 2 | 0.03 | 0.03 | 0.03 |
| TsuGNH207 | Pear | 2 | 0.28 | 0.24 | 0.21 |
| TsuGNH208 | Pear | 3 | 0.45 | 0.52 | 0.46 |
| TsuGNH250 | Pear | 2 | 0.41 | 0.33 | 0.27 |
| Averages for pear markers | 2.4 | 0.46 | 0.41 | 0.35 | |
| CsFM1097 | Tea | 4 | 0.43 | 0.49 | 0.45 |
| CsFM1206 | Tea | 3 | 0.50 | 0.57 | 0.50 |
| CsFM1566 | Tea | 4 | 0.61 | 0.49 | 0.45 |
| CsFM1595 a | Tea | 5 | 0.62 | 0.63 | 0.57 |
| MSE0348 | Tea | 5 | 0.71 | 0.61 | 0.56 |
| MSE0354 a | Tea | 4 | 0.64 | 0.80 | 0.57 |
| TM043 a | Tea | 5 | 0.45 | 0.46 | 0.40 |
| TM107 | Tea | 6 | 0.80 | 0.69 | 0.64 |
| TM336 a | Tea | 3 | 0.35 | 0.30 | 0.31 |
| TM348 | Tea | 5 | 0.64 | 0.59 | 0.51 |
| TM350 a | Tea | 4 | 0.36 | 0.36 | 0.33 |
| TM464 a | Tea | 7 | 0.51 | 0.57 | 0.45 |
| TM485 | Tea | 3 | 0.39 | 0.35 | 0.30 |
| TM553 | Tea | 3 | 0.80 | 0.50 | 0.39 |
| TM626 | Tea | 4 | 0.55 | 0.46 | 0.41 |
| Averages for tea markers | 4.3 | 0.56 | 0.52 | 0.46 |
| Marker | Plant | Average Depth (Number of Reads) | Average Depth in Replicate Analysis | Difference in Size from the Alleles in PCR-CE (bp) | Result in SSR-GBS |
|---|---|---|---|---|---|
| CH01b09b | Apple | 6834 | 5724 | 1 | Errors due to stutter band in ‘Jonathan’ and ‘Sensyu’ |
| Hi08h08 | Apple | 342 | 470 | 1 | Comparable to the result of PCR-CE |
| Hi09F01 | Apple | 395 | 605 | −4 | Comparable to the result of PCR-CE |
| Hi15h12 | Apple | 5329 | 4331 | 0 | Comparable to the result of PCR-CE |
| Hi22d06 | Apple | 3611 | 4070 | 3 | Comparable to the result of PCR-CE |
| Mdo.chr1.18 | Apple | 1206 | 1108 | −2 | Comparable to the result of PCR-CE |
| NZmsCN943067 | Apple | 1777 | 994 | −3 | Comparable to the result of PCR-CE |
| NZmsEB116209 | Apple | 747 | 534 | 3 | Comparable to the result of PCR-CE |
| NZmsEB119405 | Apple | 2442 | 2481 | 1 | Allele dropout was observed in ‘JM1’, ‘JM2’, ‘JM5’, ‘JM8’ |
| NZmsEB146613 | Apple | 8023 | 4636 | 1 | Comparable to the result of PCR-CE |
| SAmsCN944528 | Apple | 6249 | 4003 | −1 | Comparable to the result of PCR-CE |
| SAmsEB132187 | Apple | 6621 | 4034 | −3 | Comparable to the result of PCR-CE |
| TsuGNH124 | Pear | 75 | 760 | −2 | Comparable to the result of PCR-CE |
| TsuGNH161 | Pear | 16,758 | 23,807 | 2 | Comparable to the result of PCR-CE |
| TsuGNH164 | Pear | 716 | 1682 | −3 | The read depths were too low to determine genotype in ‘Oushuu’ |
| TsuGNH179 | Pear | 1671 | 4622 | −3 | Comparable to the result of PCR-CE |
| TsuGNH184 | Pear | 21 | 116 | −1 | The read depths were too low to determine genotypes |
| TsuGNH194 | Pear | 25,406 | 12,843 | 1 | Comparable to the result of PCR-CE |
| TsuGNH204 | Pear | 3877 | 1957 | −2 | Comparable to the result of PCR-CE |
| TsuGNH207 | Pear | 2718 | 1231 | 2 | Comparable to the result of PCR-CE |
| TsuGNH208 | Pear | 7348 | 3735 | −1 | Comparable to the result of PCR-CE |
| TsuGNH250 | Pear | 5021 | 2847 | −2 | Comparable to the result of PCR-CE |
| CsFM1097 | Tea | 1217 | 1868 | −4 | Comparable to the result of PCR-CE |
| CsFM1206 | Tea | 1259 | 1396 | −2 | Comparable to the result of PCR-CE |
| CsFM1566 | Tea | 857 | 1174 | −1 | Comparable to the result of PCR-CE |
| CsFM1595 | Tea | 195 | 268 | 0 | The read depths were too low to determine genotype in ‘Cha Chukanbohon No. 6’ |
| MSE0348 | Tea | 3580 | 2559 | 0 | Comparable to the result of PCR-CE |
| MSE0354 | Tea | 10,510 | 8783 | 1 | Allele dropout was observed in some varieties |
| TM043 | Tea | 3469 | 2528 | 0 | Allele dropout was observed in some varieties |
| TM107 | Tea | 2249 | 1645 | −3 | Comparable to the result of PCR-CE |
| TM336 | Tea | 14 | 12 | 1 | The read depths were too low to determine genotypes |
| TM348 | Tea | 691 | 577 | −2 | Comparable to the result of PCR-CE |
| TM350 | Tea | 514 | 357 | −3 | Allele dropout was observed in some varieties |
| TM464 | Tea | 546 | 371 | 0 | Allele dropout was observed in some varieties |
| TM485 | Tea | 2365 | 2947 | −2 | Comparable to the result of PCR-CE |
| TM553 | Tea | 6800 | 8285 | 0 | Comparable to the result of PCR-CE |
| TM626 | Tea | 1605 | 2288 | 0 | Comparable to the result of PCR-CE |
| Marker | Plant | Minimum Value of α | Optimum Value of α | Maximum Value of α | Correlation of First-Allele Frequency between Two Replicates |
|---|---|---|---|---|---|
| Hi08h08 | Apple | 0.45 | 0.57 | 0.69 | 0.98 |
| Hi09F01 | Apple | 0.69 | 0.80 | 0.90 | 0.98 |
| Hi15h12 | Apple | 0.34 | 0.43 | 0.52 | 0.98 |
| Hi22d06 | Apple | 0.57 | 0.74 | 0.90 | 1.00 |
| Mdo.chr1.18 | Apple | 0.60 | 0.61 | 0.61 | 0.98 |
| NZmsCN943067 | Apple | 0.57 | 0.66 | 0.74 | 0.99 |
| NZmsEB116209 | Apple | 0.55 | 0.70 | 0.84 | 0.99 |
| NZmsEB146613 | Apple | 0.58 | 0.73 | 0.87 | 1.00 |
| SAmsCN944528 | Apple | 0.64 | 0.78 | 0.91 | 0.99 |
| SAmsEB132187 | Apple | 0.76 | 0.82 | 0.88 | 1.00 |
| TsuGNH124 | Pear | 0.52 | 0.60 | 0.67 | 0.97 |
| TsuGNH161 | Pear | 0.53 | 0.73 | 0.93 | 1.00 |
| TsuGNH179 | Pear | 0.89 | 0.93 | 0.97 | 0.99 |
| TsuGNH194 | Pear | 0.53 | 0.75 | 0.96 | 1.00 |
| TsuGNH204 | Pear | 0.43 | 0.62 | 0.80 | 0.99 |
| TsuGNH207 | Pear | 0.53 | 0.75 | 0.97 | 1.00 |
| TsuGNH208 | Pear | 0.87 | 0.89 | 0.91 | 0.99 |
| TsuGNH250 | Pear | 0.53 | 0.75 | 0.97 | 1.00 |
| CsFM1097 | Tea | 0.55 | 0.71 | 0.87 | 0.99 |
| CsFM1206 | Tea | 0.63 | 0.77 | 0.90 | 1.00 |
| CsFM1566 | Tea | 0.71 | 0.83 | 0.95 | 0.99 |
| MSE0348 | Tea | 0.73 | 0.85 | 0.97 | 0.99 |
| TM107 | Tea | 0.72 | 0.85 | 0.98 | 0.98 |
| TM348 | Tea | 0.61 | 0.77 | 0.93 | 0.99 |
| TM485 | Tea | 0.72 | 0.84 | 0.96 | 1.00 |
| TM553 | Tea | 0.87 | 0.92 | 0.97 | 1.00 |
| TM626 | Tea | 0.73 | 0.85 | 0.97 | 1.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishio, S.; Kunihisa, M.; Taniguchi, F.; Kajiya-Kanegae, H.; Moriya, S.; Takeuchi, Y.; Sawamura, Y. Development of SSR Databases Available for Both NGS and Capillary Electrophoresis in Apple, Pear and Tea. Plants 2021, 10, 2796. https://doi.org/10.3390/plants10122796
Nishio S, Kunihisa M, Taniguchi F, Kajiya-Kanegae H, Moriya S, Takeuchi Y, Sawamura Y. Development of SSR Databases Available for Both NGS and Capillary Electrophoresis in Apple, Pear and Tea. Plants. 2021; 10(12):2796. https://doi.org/10.3390/plants10122796
Chicago/Turabian StyleNishio, Sogo, Miyuki Kunihisa, Fumiya Taniguchi, Hiromi Kajiya-Kanegae, Shigeki Moriya, Yukie Takeuchi, and Yutaka Sawamura. 2021. "Development of SSR Databases Available for Both NGS and Capillary Electrophoresis in Apple, Pear and Tea" Plants 10, no. 12: 2796. https://doi.org/10.3390/plants10122796
APA StyleNishio, S., Kunihisa, M., Taniguchi, F., Kajiya-Kanegae, H., Moriya, S., Takeuchi, Y., & Sawamura, Y. (2021). Development of SSR Databases Available for Both NGS and Capillary Electrophoresis in Apple, Pear and Tea. Plants, 10(12), 2796. https://doi.org/10.3390/plants10122796