
Detection of Persistent Viruses by High-Throughput Sequencing in Tomato and Pepper from Panama: Phylogenetic and Evolutionary Studies

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. High-Throughput Sequencing of Small RNAs
2.2. Genetic Diversity
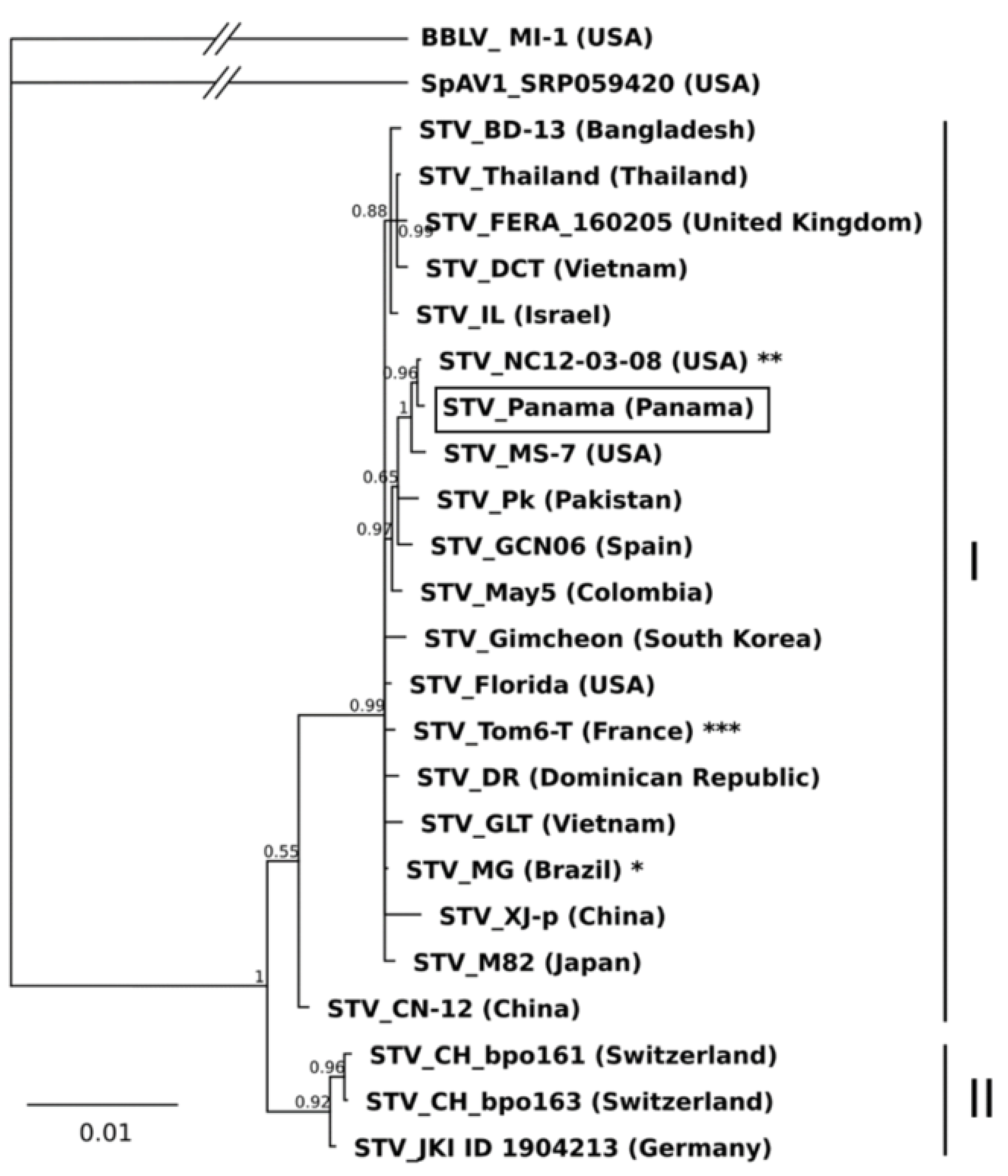
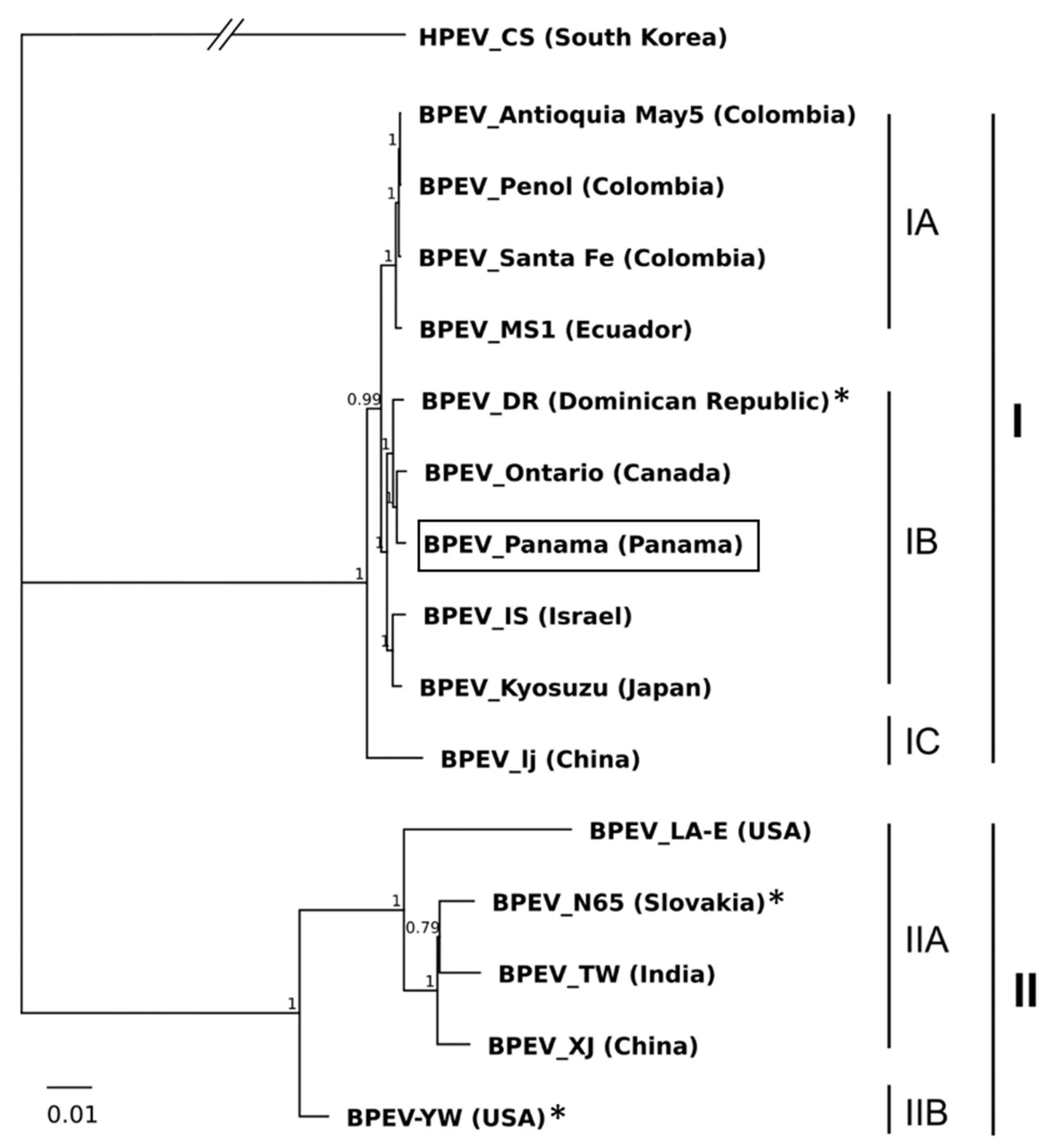
2.3. Phylogenetic Analysis
2.4. Natural Selection and Recombination Analysis
3. Materials and Methods
3.1. Plant Material, Sample Preparation and High-Throughput Sequencing
3.2. Virus Detection by RT-qPCR Assay and Sanger Sequencing
3.3. Sequence Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Safari, M.; Roossinck, M.J. Coevolution of a Persistent Plant Virus and Its Pepper Hosts. Mol. Plant-Microbe Interact. 2018, 31, 766–776. [Google Scholar] [CrossRef] [Green Version]
- Márquez, L.M.; Redman, R.S.; Rodriguez, R.J.; Roossinck, M.J. A Virus in a Fungus in a Plant: Three-Way Symbiosis Required for Thermal Tolerance. Science 2007, 315, 513–515. [Google Scholar] [CrossRef] [Green Version]
- Roossinck, M.J. The good viruses: Viral mutualistic symbioses. Nat. Rev. Genet. 2011, 9, 99–108. [Google Scholar] [CrossRef] [PubMed]
- González, L.E.; Peiró, R.; Rubio, L.; Galipienso, L. Persistent southern tomato virus (STV) interacts with cucumber mosaic and/or pepino mosaic virus in mixed-infections modifying plant symptoms, viral titer and small RNA accumulation. Microorganisms 2021, 9, 689. [Google Scholar] [CrossRef]
- Elvira-González, L.; Medina, V.; Rubio, L.; Galipienso, L. The persistent southern tomato virus modifies miRNA expression without inducing symptoms and cell ultra-structural changes. Eur. J. Plant Pathol. 2019, 156, 615–622. [Google Scholar] [CrossRef]
- Sabanadzovic, S.; Valverde, R.A.; Brown, J.K.; Martin, R.R.; Tzanetakis, I.E. Southern tomato virus: The link between the families Totiviridae and Partitiviridae. Virus Res. 2009, 140, 130–137. [Google Scholar] [CrossRef]
- Puchades, A.; Carpino, C.; Alfaro-Fernandez, A.; Font-San-Ambrosio, M.; Davino, S.; Guerri, J.; Rubio, L.; Galipienso, L. Detection of southern tomato virus by molecular hybridisation. Ann. Applied Biol. 2017, 171, 172–178. [Google Scholar] [CrossRef]
- Elvira-González, L.; Puchades, A.; Carpino, C.; Alfaro-Fernández, A.; Font-San-Ambrosio, M.; Rubio, L.; Galipienso, L. Fast detection of Southern tomato virus by one-step transcription loop-mediated isothermal amplification (RT-LAMP). J. Virol. Methods 2017, 241, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Valverde, R.A.; Khalifa, M.E.; Okada, R.; Fukuhara, T.; Sabanadzovic, S. ICTV Report Consortium ICTV Virus Taxonomy Profile: Endornaviridae. J. Gen. Virol. 2019, 100, 1204–1205. [Google Scholar] [CrossRef] [PubMed]
- Okada, R.; Kiyota, E.; Sabanadzovic, S.; Moriyama, H.; Fukuhara, T.; Saha, P.; Roossinck, M.J.; Severin, A.; Valverde, R.A. Bell pepper endornavirus: Molecular and biological properties, and occurrence in the genus Capsicum. J. Gen. Virol. 2011, 92, 2664–2673. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant Virus Metagenomics: Advances in Virus Discovery. Phytopathology 2015, 105, 716–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolja, V.V.; Krupovic, M.; Koonin, E.V. Deep Roots and Splendid Boughs of the Global Plant Virome. Annu. Rev. Phytopathol. 2020, 58, 23–53. [Google Scholar] [CrossRef]
- FAO. Statistics Division Production and Trade Statistics; FAOSTAT: Rome, Italy, 2019. [Google Scholar]
- MIDA. Agricultural Department from Government of Panama; MIDA: Panama, Panama, 2021. [Google Scholar]
- Rubio, L.; Galipienso, L.; Ferriol, I. Detection of Plant Viruses and Disease Management: Relevance of Genetic Diversity and Evolution. Front. Plant Sci. 2020, 11, 1092. [Google Scholar] [CrossRef]
- Wu, Q.; Luo, Y.; Lu, R.; Lau, N.; Lai, E.C.; Li, W.X.; Ding, S.W. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 1606–1611. [Google Scholar] [CrossRef] [Green Version]
- Santala, J.; Valkonen, J.P.T. Sensitivity of Small RNA-Based Detection of Plant Viruses. Front. Microbiol. 2018, 9, 939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elvira-González, L.; Rubio, L.; Galipienso, L. Geographically distant isolates of the persistent southern tomato virus (STV) show very low genetic diversity in the putative coat protein gene. Virus Genes 2020, 56, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in hmans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [PubMed]
- Tomašechová, J.; Hancinský, R.; Predajna, L.; Kraic, J.; Mihálik, D.; Šoltys, K.; Vávrová, S.; Böhmer, M.; Sabanadzovic, S.; Glasa, M. High-Throughput Sequencing reveals bell pepper endornavirus infection in pepper (Capsicum annum) in Slovakia and enables its further molecular characterization. Plants 2020, 9, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabanadzovic, S.; Wintermantel, W.; Valverde, R.A.; McCreight, J.D.; Ghanem-Sabanadzovic, N.A. Cucumis melo endornavirus: Genome organization, host range and co-divergence with the host. Virus Res. 2016, 214, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamilo, P.; Bianchi, N.O. Evolution of the Zfx and Zfy genes: Rates and interdependence between the genes. Mol. Biol. Evol. 1993, 10, 271. [Google Scholar] [PubMed] [Green Version]
- Adams, M.J.; Antoniw, J.F. Codon usage bias amongst plant viruses. Arch. Virol. 2003, 149, 113–135. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Pond, S.L.K.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Arenal, F.; Fraile, A.; Malpica, J.M. Variability and genetic structure of plant virus populations. Annu. Rev. Phytopathol. 2001, 39, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Falgueras, J.; Lara, A.J.; Fernández-Pozo, N.; Cantón, F.R.; Pérez-Trabado, G.; Claros, M.G. SeqTrim: A high-throughput pipeline for pre-processing any type of sequence read. BMC Bioinform. 2010, 11, 38. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.-S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elvira-González, L.; Carpino, C.; Alfaro-Fernández, A.; Ambrosio, M.I.F.-S.; Peiró, R.; Rubio, L.; Galipienso, L. A sensitive real-time RT-PCR reveals a high incidence of Southern tomato virus (STV) in Spanish tomato crops. Span. J. Agric. Res. 2018, 16, e1008. [Google Scholar] [CrossRef] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogeny. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K.; Battistuzzi, F.U. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Pond, S.L.K. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Sample | Locality | Coordinates | Host | Symptoms |
|---|---|---|---|---|
| 1 | Palma Real (Potrerillo, Dolega, Chiriquí) | 8°39′49′′ N 82°31′12′′ W | S. lycopersicum | Plant stunting, chlorosis and brown necrosis |
| 2 | El Ejido (El Ejido, Los Santos) | 7°54′18′′ N 80°22′03′′ W | C. annuum | Leaf deformation and curling |
| 3 | Tierra Blanca (El Espinal, Guararé, Los Santos) | 7°50′11′′ N 80°20′23′′ W | C. annuum | Leaf curling, mosaic and brown necrosis |
| 4 | San Ramón (Los Naranjos, Boquete, Chiriquí) | 8°48′47′′ N 82°27′42′′ W | C. annuum | Leaf mosaic and black spotted necrosis |
| Event | Position | Isolate | Major Parent | Minor Parent | Method * |
|---|---|---|---|---|---|
| 1 | 4860–5570 | BPEV-YW (JN019858) | BPEV_N65 (MN580384) | BPEV_MS1 (MN175323) | R, G, B, M, C, S, 3S |
| 2 | 6350–7162 | BPEV-YW (JN019858) | BPEV_XJ (MH182675) | BPEV_MS1 (MN175323) | R, G, B, M, C, S, 3S |
| 3 | 24–286 | BPEV-YW (JN019858) | BPEV_N65 (MN580384) | BPEV_Ontario (KT149366) | R, G, B, M, C. 3S |
| 4 | 14591–14610 | BPEV-YW (JN019858) | BPEV_TW (KU923756) | BPEV_Ontario (KT149366) | G, B, M, S, 3S |
| 5 | 14724–14728 | BPEV_DR (KX525267) | BPEV_Ontario (KT149366) | Unknown | G, B, M, 3S |
| 6 | 14662–14756 | BPEV_N65 (MN580384) | BPEV_XJ (MH182675) | BPEV_lj (KF709944) | G, B, 3S |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galipienso, L.; Elvira-González, L.; Velasco, L.; Herrera-Vásquez, J.Á.; Rubio, L. Detection of Persistent Viruses by High-Throughput Sequencing in Tomato and Pepper from Panama: Phylogenetic and Evolutionary Studies. Plants 2021, 10, 2295. https://doi.org/10.3390/plants10112295
Galipienso L, Elvira-González L, Velasco L, Herrera-Vásquez JÁ, Rubio L. Detection of Persistent Viruses by High-Throughput Sequencing in Tomato and Pepper from Panama: Phylogenetic and Evolutionary Studies. Plants. 2021; 10(11):2295. https://doi.org/10.3390/plants10112295
Chicago/Turabian StyleGalipienso, Luis, Laura Elvira-González, Leonardo Velasco, José Ángel Herrera-Vásquez, and Luis Rubio. 2021. "Detection of Persistent Viruses by High-Throughput Sequencing in Tomato and Pepper from Panama: Phylogenetic and Evolutionary Studies" Plants 10, no. 11: 2295. https://doi.org/10.3390/plants10112295
APA StyleGalipienso, L., Elvira-González, L., Velasco, L., Herrera-Vásquez, J. Á., & Rubio, L. (2021). Detection of Persistent Viruses by High-Throughput Sequencing in Tomato and Pepper from Panama: Phylogenetic and Evolutionary Studies. Plants, 10(11), 2295. https://doi.org/10.3390/plants10112295