
Integrating Genomic and Phenomic Approaches to Support Plant Genetic Resources Conservation and Use

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
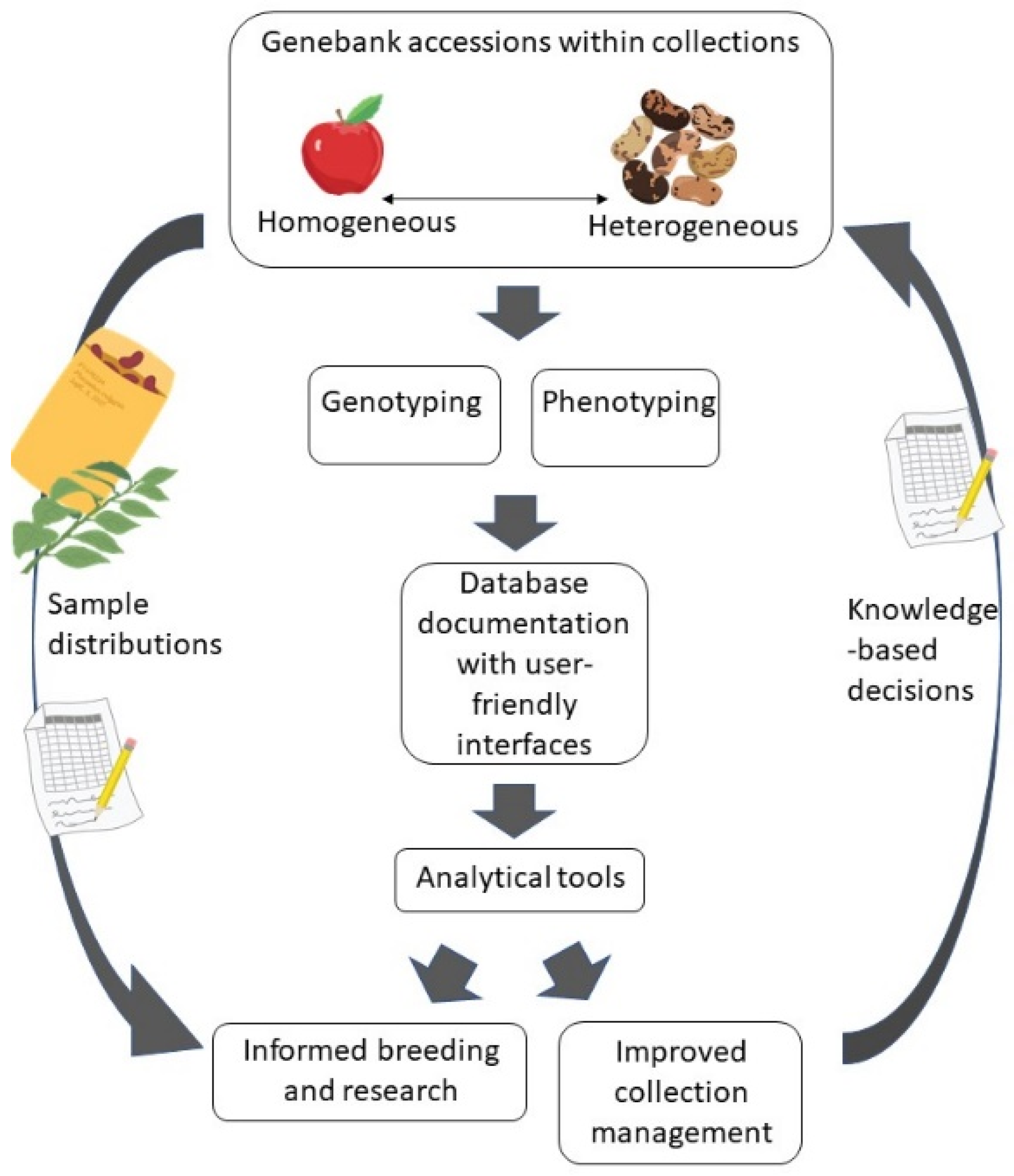
:1. Introduction
2. Customer and Stakeholder Needs for Plant Genetic Resources
3. Genomic and Phenomic Approaches Help Meet Customer and Stakeholder Needs for Plant Genetic Resources
3.1. Genomic Tools for Elucidating Germplasm Relationships
3.1.1. Germplasm Enhancement of Maize (GEM) Program for Pre-Breeding
3.1.2. Subsets for Allele Mining
3.1.3. Gene Discovery—GWAS Using Genebank Collections
3.2. Phenomics in Applied Breeding
4. Genebank Curation Needs
5. Genomic and Phenomic Approaches Help Meet Curation Needs
5.1. Genomic Data Improve Collection Management
5.2. Phenomic Data Improve Collection Use
6. Data Integration for Plant Genetic Resource Curation and Use
7. Future Prospects
Funding
Acknowledgments
Conflicts of Interest
References
- Tanksley, S.D.; McCouch, S.R. Seed banks and molecular maps: Unlocking genetic potential from the wild. Science 1997, 277, 1063–1066. [Google Scholar] [CrossRef] [Green Version]
- McCouch, S.; Baute, G.J.; Bradeen, J.; Bramel, P.; Bretting, P.K.; Buckler, E.; Burke, J.M.; Charest, D.; Cloutier, S.; Cole, G.; et al. Feeding the future. Nature 2013, 499, 23–24. [Google Scholar] [CrossRef]
- McCouch, S.; Navabi, K.; Abberton, M.; Anglin, N.L.; Barbieri, R.L.; Baum, M.; Bett, K.; Booker, H.; Brown, G.L.; Bryan, G.J.; et al. Mobilizing crop biodiversity. Mol. Plant. 2020, 13, 1341–1344. [Google Scholar] [CrossRef]
- Richards, C.M.; Lockwood, D.R.; Volk, G.M.; Walters, C. Modeling demographics and genetic diversity in ex situ collections during seed storage and regeneration. Crop Sci. 2010, 50, 2440–2447. [Google Scholar] [CrossRef]
- Ebert, A.W. The role of vegetable genetic resources in nutrition security and vegetable breeding. Plants 2020, 9, 736. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Nickson, T.E.; Challender, M. Germplasm exchange is critical to conservation of biodiversity and global food security. Agron. J. 2021, 113, 2969–2979. [Google Scholar] [CrossRef]
- Grogan, S.M.; Brown-Guedira, G.; Haley, S.D.; McMaster, G.S.; Reid, S.D.; Smith, J.; Byrne, P.F. Allelic variation in developmental genes and effects on winter wheat heading date in the U.S. Great Plains. PLoS ONE 2016, 11, e0152852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosyara, U.; Kishii, M.; Payne, T.; Sansaloni, C.P.; Singh, R.P.; Braun, H.J.; Dreisigacker, S. Genetic contribution of synthetic hexaploid wheat to CIMMYT’s spring bread wheat breeding germplasm. Sci. Rep. 2019, 9, 12355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaffney, J.; Tibebu, R.; Bart, R.; Beyene, G.; Girma, D.; Ardo Kane, N.; Mace, E.S.; Mockler, T.; Nickson, T.E.; Taylor, N.; et al. Open access to genetic sequence data maximizes value to scientists, farmers, and society. Global Food Secur. 2020, 26, 100411. [Google Scholar] [CrossRef]
- Tao, Y.; Luo, H.; Xu, J.; Cruickshank, A.; Zhao, X.; Teng, F.; Hathorn, A.; Wu, X.; Liu, Y.; Shatte, T.; et al. Extensive variation within the pan-genome of cultivated and wild sorghum. Nat. Plants 2021, 7, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Torkamaneh, D.; Lemay, M.-A.; Belzile, F. The pan-genome of the cultivated soybean (PanSoy) reveals an extraordinarily conserved gene content. Plant Biotechnol. J. 2021, 19, 1852–1862. [Google Scholar] [CrossRef]
- Faye, J.M.; Maina, F.; Akata, E.A.; Sine, B.; Diatta, C.; Mamadou, A.; Marla, S.; Bouchet, S.; Teme, N.; Rami, J.F.; et al. A genomics resource for genetics, physiology, and breeding of West African sorghum. Plant Genome 2021, 14, e20075. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.; Kamfwa, K.; Ambachew, D.; Cichy, K. Genetic variability and genome-wide association analysis of flavor and texture in cooked beans (Phaseolus vulgaris L.). Theor. Appl. Genet. 2021, 134, 959–978. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.I.; Hu, H.; Gill, M.; Batley, J.; Edwards, D. Crop breeding for a changing climate: Integrating phenomics and genomics with bioinformatics. Theor. Appl. Genet. 2021, 134, 1677–1690. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple Genotyping-by-Sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romay, M.C.; Millard, M.J.; Glaubitz, J.C.; Peiffer, J.A.; Swarts, K.L.; Casstevens, T.M.; Elshire, R.J.; Acharya, C.B.; Mitchell, S.E.; Flint-Garcia, S.A.; et al. Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biol. 2013, 14, R55. [Google Scholar] [CrossRef] [Green Version]
- Flint-Garcia, S.; Thuillet, A.-C.; Yu, J.; Pressoir, G.; Romero, S.M.; Mitchell, S.E.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Maize association population: A high-resolution platform for quantitative trait locus dissection. Plant J. 2005, 44, 1054–1064. [Google Scholar] [CrossRef]
- McMullen, M.D.; Kresovich, S.; Villeda, H.S.; Bradbury, P.; Li, H.; Sun, Q.; Flint- Garcia, S.; Thornsberry, J.; Acharya, C.; Bottoms, C.; et al. Genetic properties of the maize nested association mapping population. Science 2009, 325, 737–740. [Google Scholar] [CrossRef] [Green Version]
- Pollak, L.M. The history and success of the public-private project on Germplasm Enhancement of Maize (GEM). Adv. Agron. 2003, 78, 45–87. [Google Scholar]
- Salhuana, W.; Pollak, L. Latin American Maize Project (LAMP) and Germplasm Enhancement of Maize (GEM) project: Generating useful breeding germplasm. Maydica 2006, 51, 339–355. [Google Scholar]
- Brenner, E.A.; Blanco, M.; Gardner, C.; Lübberstedt, T. Genotypic and phenotypic characterization of isogenic doubled haploid exotic introgression lines in maize. Mol Breed. 2012, 30, 1001–1016. [Google Scholar] [CrossRef]
- Vanous, A.; Gardner, C.; Blanco, M.; Martin-Schwarze, A.; Lipka, A.E.; Flint-Garcia, S.; Bohn, M.; Edwards, J.; Lübberstedt, T. Association mapping of flowering and height traits in Germplasm Enhancement of Maize Doubled Haploid (GEM-DH) lines. Plant Genome 2018, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Qing, C.; Frei, U.; Shen, Y.; Lübberstedt, T. Association mapping for root system architecture traits under two nitrogen conditions in germplasm enhancement of maize doubled haploid lines. Crop J. 2020, 8, 213–226. [Google Scholar] [CrossRef]
- Vanous, A.; Gardner, C.; Blanco, M.; Martin-Schwarze, A.; Wang, J.; Li, X.; Lipka, A.E.; Flint-Garcia, S.; Bohn, M.; Edwards, J.; et al. Stability analysis of kernel quality traits in exotic-derived doubled haploid maize lines. Plant Genome 2019, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Frankel, O.H. Genetic perspectives of germplasm conservation. In Genetic Manipulation: Impact on Man and Society; Arber, W., Illemensee, K., Peacock, W.J., Starlinger, P., Eds.; Cambridge University Press: Cambridge, UK, 1984; pp. 161–170. [Google Scholar]
- Brown, A. The case for core collections. In The Use of Plant Genetic Resources; Brown, A.H.D., Frankel, O.H., Marshall, D.R., Williams, J.T., Eds.; Cambridge University Press: Cambridge, UK, 1989; pp. 136–156. [Google Scholar]
- Zila, C.T.; Samayoa, L.F.; Santiago, R.; Butrón, A.; Holland, J.B. A genome-wide association study reveals genes associated with Fusarium ear rot resistance in a Maize Core Diversity Panel. G3 Genes|Genomes|Genetics 2013, 3, 2095–2104. [Google Scholar] [CrossRef] [Green Version]
- Peiffer, J.A.; Romay, M.C.; Gore, M.A.; Flint-Garcia, S.A.; Zhang, Z.; Millard, M.J.; Gardner, C.A.C.; McMullen, M.D.; Holland, J.B.; Bradbury, P.J.; et al. The genetic architecture of maize height. Genetics 2014, 196, 1337–1356. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Romay, M.C.; Glaubitz, J.C.; Bradbury, P.J.; Elshire, R.J.; Wang, T.; Li, Y.; Li, Y.; Semagn, K.; Zhang, X.; et al. High-resolution genetic mapping of maize pan-genome sequence anchors. Nat. Commun. 2015, 6, 6914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, S.; Bradbury, P.J.; Casstevens, T.; Holland, J.B. Genetic architecture of domestication-related traits in maize. Genetics 2016, 204, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansey, C.N.; Johnson, J.M.; Sekhon, R.S.; Kaeppler, S.M.; de Leon, N. Genetic diversity of a maize association population with restricted phenology. Crop Sci. 2011, 51, 704–715. [Google Scholar] [CrossRef]
- Gage, J.L.; White, M.R.; Edwards, J.W.; Kaeppler, S.; de Leon, N. Selection signatures underlying dramatic male inflorescence transformation during modern hybrid maize breeding. Genetics 2018, 210, 1125–1138. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, T.; Leon, N.; Kaeppler, S.; Tracy, W. Genetic analysis of resistance in the Wisconsin Diversity Panel of maize. Crop Sci. 2018, 58, 1853–1865. [Google Scholar] [CrossRef]
- Mazaheri, M.; Heckwolf, M.; Vaillancourt, B.; Gage, J.L.; Burdo, B.; Heckwolf, S.; Barry, K.; Lipzen, A.; Ribeiro, C.B.; Kono, T.J.Y.; et al. Genome-wide association analysis of stalk biomass and anatomical traits in maize. BMC Plant Biol. 2019, 19, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, C.H.; Qiu, Y.; Coletta, R.D.; Renk, J.S.; Monnahan, P.J.; Noshay, J.M.; Gilbert, A.; Anderson, A.N.; McGaugh, S.E.; Springer, N.M.; et al. Population level variation of transposable elements in a Maize Diversity Panel. bioRxiv 2020. [Google Scholar] [CrossRef]
- Renk, J.S.; Gilbert, A.M.; Hattery, T.J.; O’Connor, C.H.; Monnahan, P.J.; Anderson, N.; Waters, A.J.; Eickholt, D.; Flint-Garcia, S.A.; Yandeau-Nelson, M.D.; et al. Genetic architecture of kernel compositional variation in a maize diversity panel. Plant Genome 2021, in press. Available online: https://www.biorxiv.org/content/10.1101/2021.03.29.436703v1.full (accessed on 20 October 2021). [CrossRef]
- Burns, M.J.; Renk, J.S.; Eickholt, D.P.; Gilbert, A.M.; Hattery, T.J.; Holmes, M.; Anderson, N.; Waters, A.J.; Kalambur, S.; Flint-Garcia, S.A.; et al. Predicting moisture content during maize nixtamalization using machine learning with NIR spectroscopy. Theor. Appl. Genet. 2021, 134, 3743–3757. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.; Eshed, R.; Rozen, A.; Zviran, T.; Kuhn, D.N.; Irihimivitch, V.; Sherman, A.; Ophir, R. Genetic diversity of avocado (Persea americana Mill.) germplasm using pooled sequencing. BMC Genom. 2019, 20, 379. [Google Scholar] [CrossRef]
- Schlötterer, C.; Tobler, R.; Kofler, R.; Nolte, V. Sequencing pools of individuals—Mining genome wide polymorphism data without big funding. Nat. Genet. 2014, 15, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Harloff, H.J.; Melzer, S.; Leineweber, J.; Defant, B.; Jung, C. A rhomboid like protease gene from an interspecies translocation confers resistance to cyst nematodes. New Phytol. 2021, 231, 801–813. [Google Scholar] [CrossRef]
- Danilevicz, M.F.; Fernandez, C.G.T.; Marsh, J.I.; Bayer, P.E.; Edwards, D. Plant pangenomics: Approaches, applications and advancements. Curr. Opin. Plant Biol. 2020, 54, 18–25. [Google Scholar] [CrossRef]
- Galewski, P.; McGrath, J.M. Genetic diversity among cultivated beets (Beta vulgaris) assessed via population based whole genome sequences. BMC Genom. 2020, 21, 189. [Google Scholar] [CrossRef] [Green Version]
- Lv, Q.; Li, W.; Sun, Z.; Ouyang, N.; Jing, X.; He, Q.; Wu, J.; Zheng, J.; Zheng, J.; Tang, S.; et al. Resequencing of 1,143 indica rice accessions reveals important genetic variations and different heterosis patterns. Nat. Commun. 2020, 11, 4778. [Google Scholar] [CrossRef] [PubMed]
- Torkamaneh, D.; Laroche, J.; Valliyodan, B.; O’Donoughue, L.; Cober, E.; Rajcan, I.; Vilela Abdelnoor, R.; Sreedasyam, A.; Schmutz, J.; Nguyen, H.T.; et al. Soybean (Glycine max) Haplotype Map (GmHapMap): A universal resource for soybean translational and functional genomics. Plant Biotechnol. J. 2021, 19, 324–334. [Google Scholar] [CrossRef]
- Varshney, R.K.; Thudi, M.; Roorkiwal, M.; He, W.; Upadhyaya, H.D.; Yang, W.; Bajaj, P.; Cubry, P.; Rathore, A.; Jian, J.; et al. Resequencing of 429 chickpea accessions from 45 countries provides insights into genome diversity, domestication and agronomic traits. Nat. Genet. 2019, 51, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Tibbs Cortes, L.; Zhang, Z.; Yu, J. Status and prospects of genome-wide association studies in plants. Plant Genome 2021, 14, e20077. [Google Scholar] [CrossRef] [PubMed]
- Crossa, J. From genotype × environment interaction to gene × environment interaction. Curr. Genom. 2012, 13, 225–244. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.S. Using genotype-by-environment interaction for crop cultivar development. Adv. Agron. 1997, 62, 199–252. [Google Scholar] [CrossRef]
- Sandhu, K.S.; Mihalyov, P.D.; Lewien, M.J.; Pumphrey, M.O.; Carter, A.H. Combining genomic and phenomic information for predicting grain protein content and grain yield in spring wheat. Front. Plant Sci. 2021, 12, 170. [Google Scholar] [CrossRef]
- Rebetzke, G.J.; Jimenez-Berni, J.; Fischer, R.A.; Deery, D.M.; Smith, D.J. High-throughput phenotyping to enhance the use of crop genetic resources. Plant Sci. 2019, 282, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Harper, L.; Campbell, J.; Cannon, E.K.; Jung, S.; Poelchau, M.; Walls, R.; Andorf, C.; Arnaud, E.; Berardini, T.Z.; Birkett, C.; et al. AgBioData consortium recommendations for sustainable genomics and genetics databases for agriculture. Database 2018, 2018, bay088. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, R.; Matteis, L.; Skofic, M.; Portugal, A.; McLaren, G.; Hyman, G.; Arnaud, E. Bridging the phenotypic and genetic data useful for integrated breeding through a data annotation using the Crop Ontology developed by the crop communities of practice. Front. Physiol. 2012, 3, 326. [Google Scholar] [CrossRef] [Green Version]
- Cooper, L.; Meier, A.; Laporte, M.A.; Elser, J.L.; Mungall, C.; Sinn, B.T.; Cavaliere, D.; Carbon, S.; Dunn, N.A.; Smith, B.; et al. The Planteome database: An integrated resource for reference ontologies, plant genomics and phenomics. Nucleic Acids Res. 2018, 46, D1168–D1180. [Google Scholar] [CrossRef] [Green Version]
- Jonquet, C.; Toulet, A.; Arnaud, E.; Aubin, S.; Yeumo, E.D.; Emonet, V.; Graybeal, J.; Laporte, M.A.; Musen, M.A.; Pesce, V.; et al. AgroPortal: A vocabulary and ontology repository for agronomy. Comput. Electron. Agric. 2018, 144, 126–143. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 10018. [Google Scholar] [CrossRef] [Green Version]
- Bretting, P.K.; Widrlechner, M. Genetic markers and plant genetic resource management. Plant Breed. Rev. 1995, 13, 11–86. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.M.; Gujaria-Verma, N.; Ramsay, L.; Yuan, H.Y.; Caron, C.; Diapari, M.; Vandenberg, A.; Bett, K.E. Classification and characterization of species within the genus Lens using genotyping-by-sequencing (GBS). PLoS ONE 2015, 10, e0122025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, S.G.; Jost, M.; Taketa, S.; Mazón, E.R.; Himmelbach, A.; Oppermann, M.; Weise, S.; Knüpffer, H.; Basterrechea, M.; König, P.; et al. Genebank genomics highlights the diversity of a global barley collection. Nat. Genet. 2019, 51, 319–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, D.; Chavez, O.; Coombs, J.; Soto, J.; Gomez, R.; Douches, D.; Panta, A.; Silvestre, R.; Anglin, N.L. Genetic identity in genebanks: Application of the SolCAP 12K SNP array in fingerprinting and diversity analysis in the global in trust potato collection. Genome 2018, 61, 523–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Kumar, P.; Shi, A.; Mou, B. Population genetics and genome-wide association studies provide insights into the influence of selective breeding on genetic variation in lettuce. Plant Genome 2021, e20086. [Google Scholar] [CrossRef]
- Watts, S.; Migicovsky, Z.; McClure, K.A.; Yu, C.H.J.; Amyotte, B.; Baker, T.; Bowlby, D.; Burgher-MacLellan, K.; Butler, L.; Donald, R.; et al. Quantifying apple diversity: A phenomic characterization of Canada’s Apple Biodiversity Collection. Plants People Planet 2021. [Google Scholar] [CrossRef]
- Gross, B.L.; Volk, G.M.; Richards, C.M.; Forsline, P.L.; Fazio, G.; Thomas, C.T. Identification of “duplicate” accessions within the USDA-ARS National Plant Germplasm System Malus collection. J. Am. Soc. Hort. Sci. 2012. 137, 333–342. [CrossRef] [Green Version]
- Gottschalk, C.; (USDA-ARS, Kearneysville, WV, USA). Personal communication, 2021.
- Yu, X.; Li, X.; Guo, T.; Zhu, C.; Wu, Y.; Mitchell, S.E.; Roozeboom, K.L.; Wang, D.; Wang, M.L.; Pederson, G.A.; et al. Genomic prediction contributing to a promising global strategy to turbocharge gene banks. Nat. Plants 2016. 2, 16150. [CrossRef]
- Sankaran, S.; Mishra, A.; Ehsani, R.; Davis, C. A review of advanced techniques for detecting plant diseases. Comput. Electron. Agric. 2010, 72, 1–13. [Google Scholar] [CrossRef]
- Sangjan, W.; Sankaran, S. Phenotyping architecture traits of tree species using remote sensing techniques. Trans. ASABE 2021, in press. [Google Scholar] [CrossRef]
- Zhang, C.; Craine, W.A.; McGee, R.J.; Vandemark, G.J.; Davis, J.B.; Brown, J.; Hulbert, S.H.; Sankaran, S. High-throughput phenotyping of canopy height in cool-season crops using sensing techniques. Agron. J. 2021, 113, 3269–3280. [Google Scholar] [CrossRef]
- Zhang, Z.; (Department of Crop and Soil Science, Washington State University, Pullman, WA, USA). Personal communication, 2021.
- Sankaran, S.; Khot, L.R.; Espinoza, C.Z.; Jarolmasjed, S.; Sathuvalli, V.R.; Vandemark, G.J.; Miklas, P.N.; Carter, A.H.; Pumphrey, M.O.; Knowles, N.R.; et al. Low-altitude, high-resolution aerial imaging systems for row and field crop phenotyping: A review. Eur. J. Agron. 2015, 70, 112–123. [Google Scholar] [CrossRef]
- Andrade-Sanchez, P.; Gore, M.A.; Heun, J.T.; Thorp, K.R.; Carmo-Silva, A.E.; French, A.N.; Salvucci, M.E.; White, J.W. Development and evaluation of a field-based high-throughput phenotyping platform. Funct. Plant Biol. 2013, 41, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, K.M. Using UAV-Based Imagery to Determine Volume, Groundcover, and Growth Rate Characteristics of Lentil (Lens culinaris Medik.). Ph.D. Thesis, University of Saskatchewan, Saskatoon, Saskatchewan, 2020. [Google Scholar]
- Crain, J.L.; Wei, Y.; Barker III, J.; Thompson, S.M.; Alderman, P.D.; Reynolds, M.; Zhang, N.; Poland, J. Development and deployment of a portable field phenotyping platform. Crop Sci. 2016, 56, 965–975. [Google Scholar] [CrossRef]
- Zhang, Z.; Kayacan, E.; Thompson, B.; Chowdhary, G. High precision control and deep learning-based corn stand counting algorithms for agricultural robot. Auton. Robot. 2020, 44, 1289–1302. [Google Scholar] [CrossRef]
- Wright, D.M.; Neupane, S.; Heidecker, T.; Haile, T.A.; Chan, C.; Coyne, C.J.; McGee, R.J.; Udupa, S.; Henkrar, F.; Barilli, E.; et al. Understanding photothermal interactions will help expand production range and increase genetic diversity of lentil (Lens culinaris Medik.). Plants People Planet 2021, 3, 171–181. [Google Scholar] [CrossRef]
- Silva-Díaz, C.; Ramírez, D.A.; Rinza, J.; Ninanya, J.; Loayza, H.; Gómez, R.; Anglin, N.L.; Eyzaguirre, R.; Quiroz, R. Radiation interception, conversion and partitioning efficiency in potato landraces: How far are we from the optimum? Plants 2020, 9, 787. [Google Scholar] [CrossRef]
- Nguyen, G.N.; Norton, S.L. Genebank phenomics: A strategic approach to enhance value and utilization of crop germplasm. Plants 2020, 9, 817. [Google Scholar] [CrossRef] [PubMed]
- Halcro, K.; McNabb, K.; Lockinger, A.; Socquet-Juglard, D.; Bett, K.E.; Noble, S.D. The BELT and phenoSEED platforms: Shape and colour phenotyping of seed samples. Plant Methods 2020, 16, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, J.; Lee, E.; Kim, N.; Kim, S.L.; Choi, I.; Ji, H.; Chung, Y.S.; Choi, M.S.; Moon, J.K.; Kim, K.H. High throughput phenotyping for various traits on soybean seeds using image analysis. Sensors 2020, 20, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komyshev, E.; Genaev, M.; Afonnikov, D. Evaluation of the SeedCounter, a mobile application for grain phenotyping. Front. Plant Sci. 2017, 7, 1990. [Google Scholar] [CrossRef] [Green Version]
- Makanza, R.; Zaman-Allah, M.; Cairns, J.E.; Eyre, J.; Burgueño, J.; Pacheco, Á.; Diepenbrock, C.; Magorokosho, C.; Tarekegne, A.; Olsen, M.; et al. High-throughput method for ear phenotyping and kernel weight estimation in maize using ear digital imaging. Plant Methods 2018, 14, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, N.D.; Haase, N.J.; Lee, J.; Kaeppler, S.M.; de Leon, N.; Spalding, E.P. A robust, high-throughput method for computing maize ear, cob, and kernel attributes automatically from images. Plant J. 2017, 89, 169–178. [Google Scholar] [CrossRef]
- Eizenga, G.C.; McClung, A.M.; Huggins, T.D. Registration of two Oryza sativa tropical japonica germplasm lines selected for panicle architecture and grain size traits. J. Plant Regist. 2021, 15, 573–582. [Google Scholar] [CrossRef]
- Tanabata, T.; Shibaya, T.; Hori, K.; Ebana, K.; Yano, M. SmartGrain: High-throughput phenotyping software for measuring seed shape through image analysis. Plant Physiol. 2012, 160, 1871–1880. [Google Scholar] [CrossRef] [Green Version]
- Genomics Open-Source Breeding Informatics Initiative. Available online: http://cbsugobii05.biohpc.cornell.edu/wordpress/ (accessed on 28 September 2021).
- Breeding Insight. Available online: https://breedinginsight.org/ (accessed on 29 September 2021).
- Excellence in Breeding Platform. Available online: https://excellenceinbreeding.org/ (accessed on 29 September 2021).
- Breedbase. Available online: https://breedbase.org (accessed on 29 September 2021).
- Hershberger, J.; Morales, N.; Simoes, C.C.; Ellerbrock, B.; Bauchet, G.; Mueller, L.A.; Gore, M.A. Making waves in Breedbase: An integrated spectral data storage and analysis pipeline for plant breeding programs. Plant Phenome J. 2021, 4, e20012. [Google Scholar] [CrossRef]
- Morales, N.; Bauchet, G.J.; Tantikanjana, T.; Powell, A.F.; Ellerbrock, B.J.; Tecle, I.Y.; Mueller, L.A. High density genotype storage for plant breeding in the Chado schema of Breedbase. PLoS ONE 2020, 15, e0240059. [Google Scholar] [CrossRef]
- BrAPI. The Breeding API. Available online: https://brapi.org/ (accessed on 29 September 2021).
- Pixley, K.V.; Salinas-Garcia, G.E.; Hall, A.; Kropff, M.; Ortiz, C.; Bouvet, L.; Suhalia, A.; Vikram, P.; Singh, S. CIMMYT’s Seeds of Discovery Initiative: Harnessing Biodiversity for Food Security and Sustainable Development. Indian J. Plant Genet. Resour. 2018, 31, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Seeds of Discovery. Available online: https://seedsofdiscovery.org/ (accessed on 29 September 2021).
- Agent. Global access to plant genetic resources. Available online: https://agent-project.eu/ (accessed on 29 September 2021).
- Germinate. Available online: https://germinateplatform.github.io/get-germinate/ (accessed on 29 September 2021).
- BRIDGE web portal. Available online: https://bridge.ipk-gatersleben.de/ (accessed on 29 September 2021).
- Andorf, C.M.; Cannon, E.K.; Portwood, J.L.; Gardiner, J.M.; Harper, L.C.; Schaeffer, M.L.; Braun, B.L.; Campbell, D.A.; Vinnakota, A.G.; Sribalusu, V.V.; et al. MaizeGDB update: New tools, data and interface for the maize model organism database. Nucleic Acids Res. 2016, 44, D1195–D1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MaizeGDB TypSimSelector. Available online: https://www.maizegdb.org/TYPSimSelector (accessed on 29 September 2021).
- König, P.; Beier, S.; Basterrechea, M.; Schüler, D.; Arend, D.; Mascher, M.; Stein, N.; Scholz, U.; Lange, M. BRIDGE–a visual analytics web tool for barley genebank genomics. Front. Plant Sci. 2020, 11, 701. [Google Scholar] [CrossRef] [PubMed]
- Raubach, S.; Kilian, B.; Dreher, K.; Amri, A.; Bassi, F.M.; Boukar, O.; Cook, D.; Cruickshank, A.; Fatokun, C.; El Haddad, N.; et al. From bits to bites: Advancement of the Germinate platform to support prebreeding informatics for crop wild relatives. Crop Sci. 2021, 61, 1538–1566. [Google Scholar] [CrossRef]
- Bari, A.; Street, K.; Mackay, M.; Endresen, D.T.F.; De Pauw, E.; Amri, A. Focused identification of germplasm strategy (FIGS) detects wheat stem rust resistance linked to environmental variables. Genetic Resources Crop Evol. 2012, 59, 1465–1481. [Google Scholar] [CrossRef]
- Thachuk, C.; Crossa, L.; Franco, J.; Dreisigacker, S.; Warburton, M.; Davenport, G.F. Core Hunter: An algorithm for sampling genetic resources based on multiple genetic measures. BMC Bioinform. 2009, 10, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belzile, F.; Abed, A.; Torkamaneh, D. Time for a paradigm shift in the use of plant genetic resources. Genome 2020, 63, 189–194. [Google Scholar] [CrossRef]
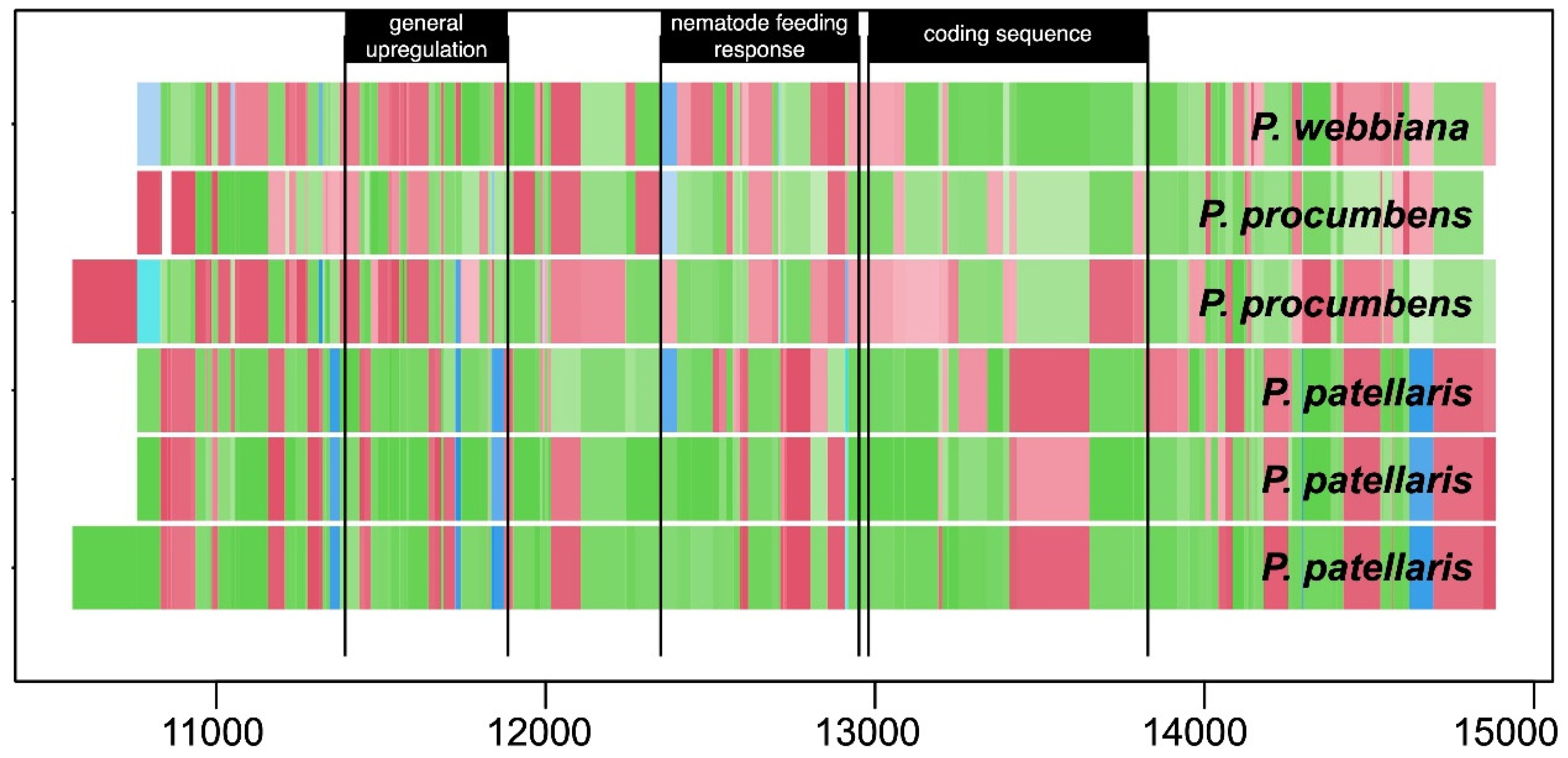
- Reeves, P.A.; Tetreault, H.M.; Richards, C.M. Bioinformatic extraction of functional genetic diversity from heterogeneous germplasm collections for crop improvement. Agronomy 2020, 10, 593. [Google Scholar] [CrossRef]
- Al Bari, M.A.; Zheng, P.; Viera, I.; Worral, H.; Szwiec, S.; Ma, Y.; Main, D.; Coyne, C.J.; McGee, R.; Bandillo, N. Harnessing genetic diversity in the USDA pea (Pisum sativum L.) germplasm collection through genomic prediction. bioRxiv 2021. [Google Scholar]
- Jiang, Y.; Weise, S.; Graner, A.; Reif, J.C. Using genome-wide predictions to assess the phenotypic variation of a barley (Hordeum sp.) gene bank collection for important agronomic traits and passport information. Front. Plant Sci. 2021, 11, 2180. [Google Scholar] [CrossRef]
- Allier, A.; Teyssèdre, S.; Lehermeier, C.; Moreau, L.; Charcosset, A. Optimized breeding strategies to harness genetic resources with different performance levels. BMC Genom. 2020, 21, 349. [Google Scholar] [CrossRef] [PubMed]
- Bayer, P.E.; Petereit, J.; Danilevicz, M.F.; Anderson, R.; Batley, J.; Edwards, D. The application of pangenomics and machine learning in genomic selection in plants. Plant Genome 2021, e20112. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volk, G.M.; Byrne, P.F.; Coyne, C.J.; Flint-Garcia, S.; Reeves, P.A.; Richards, C. Integrating Genomic and Phenomic Approaches to Support Plant Genetic Resources Conservation and Use. Plants 2021, 10, 2260. https://doi.org/10.3390/plants10112260
Volk GM, Byrne PF, Coyne CJ, Flint-Garcia S, Reeves PA, Richards C. Integrating Genomic and Phenomic Approaches to Support Plant Genetic Resources Conservation and Use. Plants. 2021; 10(11):2260. https://doi.org/10.3390/plants10112260
Chicago/Turabian StyleVolk, Gayle M., Patrick F. Byrne, Clarice J. Coyne, Sherry Flint-Garcia, Patrick A. Reeves, and Chris Richards. 2021. "Integrating Genomic and Phenomic Approaches to Support Plant Genetic Resources Conservation and Use" Plants 10, no. 11: 2260. https://doi.org/10.3390/plants10112260
APA StyleVolk, G. M., Byrne, P. F., Coyne, C. J., Flint-Garcia, S., Reeves, P. A., & Richards, C. (2021). Integrating Genomic and Phenomic Approaches to Support Plant Genetic Resources Conservation and Use. Plants, 10(11), 2260. https://doi.org/10.3390/plants10112260