Molecular and Cellular Pathogenesis of Ellis-van Creveld Syndrome: Lessons from Targeted and Natural Mutations in Animal Models


Abstract
1. Introduction
2. Primary Cilium, Ciliopathy, and EVC
3. Overview of Human Signs
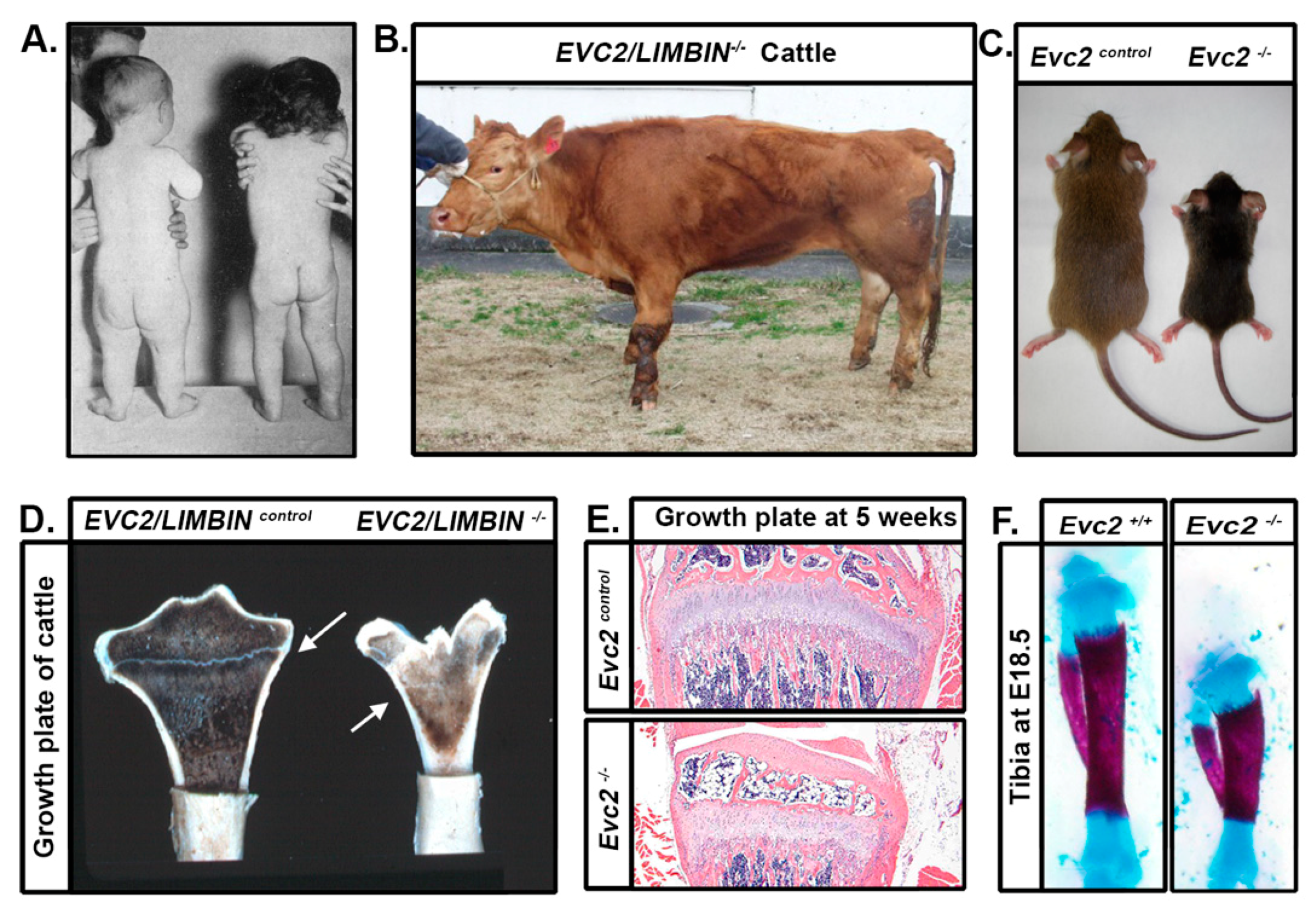
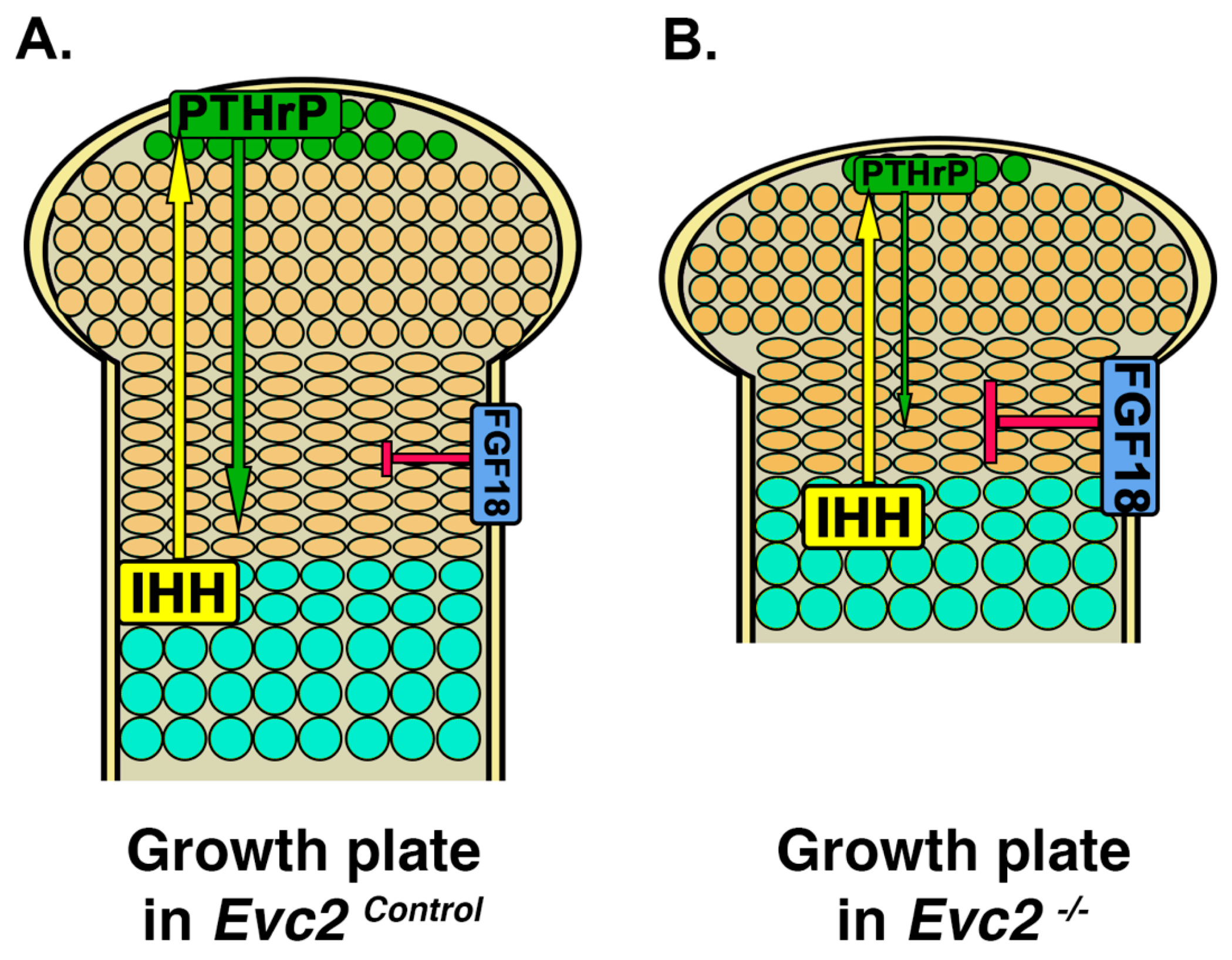
4. Limb Phenotypes
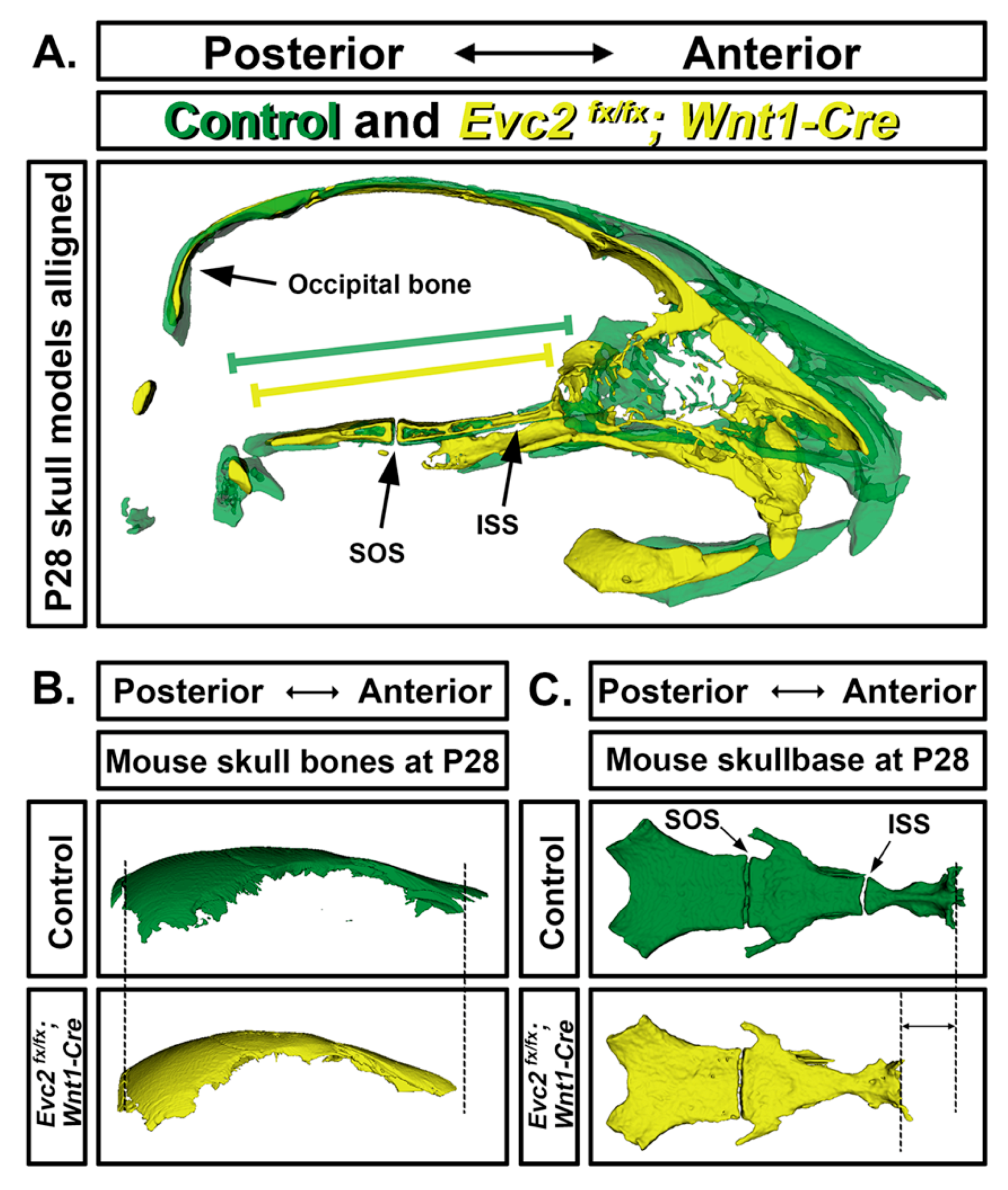
5. Craniofacial Phenotype
6. Tooth Phenotype
7. Tracheal Cartilage Phenotypes
8. EVC-Like Disorders in Other Non-Human Species
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ellis, R.W.B.; Van Creveld, S. A syndrome characterized by ectodermal dysplasia, polydactyly, chondro-dysplasia and congenital morbus cordis: Report of three cases. Arch. Dis. Child. 1940, 15, 65–84. [Google Scholar] [CrossRef] [PubMed]
- McKusick, V.A. Dwarfism in the Amish I. The Ellis-van Creveld Syndrome. Bull. Johns Hopkins Hosp. 1964, 115, 306–336. [Google Scholar] [PubMed]
- Ruiz-Perez, V.L.; Ide, S.E.; Strom, T.M.; Lorenz, B.; Wilson, D.; Woods, K.; King, L.; Francomano, C.; Freisinger, P.; Spranger, S.; et al. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat. Genet. 2000, 24, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Baujat, G.; Merrer, M.L. Ellis-Van Creveld syndrome. Orphanet J. Rare Dis. 2007, 2. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Perez, V.L.; Goodship, J.A. Ellis-van Creveld syndrome and Weyers acrodental dysostosis are caused by cilia-mediated diminished response to Hedgehog ligands. Am. J. Med. Genet. Part C Semin. Med. Genet. 2009, 151, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Tompson, S.W.; Ruiz-Perez, V.L.; Blair, H.J.; Barton, S.; Navarro, V.; Robson, J.L.; Wright, M.J.; Goodship, J.A. Sequencing EVC and EVC2 identifies mutations in two-thirds of Ellis-van Creveld syndrome patients. Hum. Genet. 2007, 120, 663–670. [Google Scholar] [CrossRef] [PubMed]
- D’Asdia, M.C.; Torrente, I.; Consoli, F.; Ferese, R.; Magliozzi, M.; Bernardini, L.; Guida, V.; Digilio, M.C.; Marino, B.; Dallapiccola, B.; et al. Novel and recurrent EVC and EVC2 mutations in Ellis-van Creveld syndrome and Weyers acrofacial dyostosis. Eur. J. Med. Genet. 2013, 56, 80–87. [Google Scholar] [CrossRef]
- Takeda, H.; Takami, M.; Oguni, T.; Tsuji, T.; Yoneda, K.; Sato, H.; Ihara, N.; Itoh, T.; Kata, S.R.; Mishina, Y.; et al. Positional cloning of the gene LIMBIN responsible for bovine chondrodysplastic dwarfism. Proc. Natl. Acad. Sci. USA 2002, 99, 10549–10554. [Google Scholar] [CrossRef]
- Zhang, H.; Takeda, H.; Tsuji, T.; Kamiya, N.; Rajderkar, S.; Louie, K.A.; Collier, C.; Scott, G.; Ray, M.; Mochida, Y.; et al. Generation of Evc2/Limbin global and conditional KO mice and its roles during mineralized tissue formation. Genesis 2015, 53, 612–626. [Google Scholar] [CrossRef]
- Ruiz-Perez, V.L.; Blair, H.J.; Rodrigues-Andres, M.E.; Blanco, M.J.; Wilson, A.; Liu, Y.N.; Miles, C.; Peters, H.; Goodship, J.A. Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development 2007, 134, 2903–2912. [Google Scholar] [CrossRef]
- Caparrós-Martín, J.A.; Valencia, M.; Reytor, E.; Pacheco, M.; Fernandez, M.; Perez-Aytes, A.; Gean, E.; Lapunzina, P.; Peters, H.; Goodship, J.A.; et al. The ciliary EVC/EVC2 complex interacts with smo and controls hedgehog pathway activity in chondrocytes by regulating Sufu/Gli3 dissociation and Gli3 trafficking in primary cilia. Hum. Mol. Genet. 2013, 22, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, W.; Chen, Y.; Jiang, J. Smoothened transduces Hedgehog signal by forming a complex with Evc/Evc2. Cell Res. 2012, 22, 1593–1604. [Google Scholar] [CrossRef]
- Eggenschwiler, J.T.; Anderson, K.V. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007, 23, 345–373. [Google Scholar] [CrossRef] [PubMed]
- Afzelius, B.A. Cilia-related diseases. J. Pathol. 2004, 204, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Camner, P.; Mossberg, B.; Afzelius, B.A. Evidence of congenitally nonfunctioning cilia in the tracheobronchial tract in two subjects. Am. Rev. Respir. Dis. 1975, 112, 807–809. [Google Scholar] [CrossRef]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef]
- Kim, J.; Kato, M.; Beachy, P.A. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. USA 2009, 106, 21666–21671. [Google Scholar] [CrossRef]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef]
- Dorn, K.V.; Hughes, C.E.; Rohatgi, R. A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev. Cell 2012, 23, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.J.; Tompson, S.; Liu, Y.N.; Campbell, J.; MacArthur, K.; Ponting, C.P.; Ruiz-Perez, V.L.; Goodship, J.A. Evc2 is a positive modulator of Hedgehog signalling that interacts with Evc at the cilia membrane and is also found in the nucleus. BMC Biol. 2011, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Valencia, M.; Lapunzina, P.; Lim, D.; Zannolli, R.; Bartholdi, D.; Wollnik, B.; Al-Ajlouni, O.; Eid, S.S.; Cox, H.; Buoni, S.; et al. Widening the mutation spectrum of EVC and EVC2: Ectopic expression of Weyer variants in NIH 3T3 fibroblasts disrupts Hedgehog signaling. Hum. Mutat. 2009, 30, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Ulucan, H.; Guel, D.; Sapp, J.C.; Cockerham, J.; Johnston, J.J.; Biesecker, L.G. Extending the spectrum of Ellis van Creveld syndrome: A large family with a mild mutation in the EVC gene. BMC Med. Genet. 2008, 9, 92. [Google Scholar] [CrossRef]
- Zhang, H.; Kamiya, N.; Tsuji, T.; Takeda, H.; Scott, G.; Rajderkar, S.; Ray, M.K.; Mochida, Y.; Allen, B.; Lefebvre, V.; et al. Elevated Fibroblast Growth Factor Signaling Is Critical for the Pathogenesis of the Dwarfism in Evc2/Limbin Mutant Mice. PLoS Genet. 2016, 12, e1006510. [Google Scholar] [CrossRef]
- Alvarez-Borja, A. Ellis-van Creveld Syndrome: Report of two cases. Pediatrics 1960, 26, 301–309. [Google Scholar] [CrossRef]
- Husson, G.S.; Parkman, P. Chondroectodermal dysplasia (Ellis-Van Creveld syndrome) with a complex cardiac malformation. Pediatrics 1961, 28, 285–292. [Google Scholar]
- Lynch, J.I. Congenital Heart Disease and Chondroectodermal Dysplasia. Am. J. Dis. Child. 1968, 115, 80. [Google Scholar] [CrossRef]
- Goor, D.; Rotem, Y.; Friedman, A.; Neufeld, H.N. ELLIS-VAN CREVELD SYNDROME IN IDENTICAL TWINS * In 1933 McIntosh (Holt and McIntosh, 1933) described abnormal findings in a girl who had. Br. Heart J. 1965, 27, 797–804. [Google Scholar] [CrossRef][Green Version]
- Metrakos, J.D.; Fraser, F.C. Evidence for a hereditary factor in chondroectodermal dysplasia (Ellis-van Creveld syndrome). Am. J. Hum. Genet. 1954, 6, 260–269. [Google Scholar]
- Smith, H.L.; Hand, A.M. Chondroectodermal Dysplasia (Ellis-van Creveld Syndrome): Report of Two Cases. Pediatrics 1958, 21, 298–307. [Google Scholar] [PubMed]
- Weiss, H.; Crosett, A.D. Chondroectodermal dysplasia: Report of a case and review of the literature. J. Pediatrics 1955, 46, 268–275. [Google Scholar] [CrossRef]
- Mitchell, F.N.; Waddell, W.W. Ellis-van Creveld syndrome: Report of two cases in siblings. Acta Paediatr. 1958, 47, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.D. Two cases of Ellis van Creveld syndrome in a small island population. J. Med. Genet. 1977, 14, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, E.O.; Janovitz, D.; Cavalcanti De Albuquerque, S. Ellis-van Creveld syndrome: Report of 15 cases in an inbred kindred. J. Med. Genet. 1980, 17, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Cahuana, A.; Palma, C.; Gonzáles, W.; Geán, E. Oral manifestations in Ellis-van Creveld syndrome: Report of five cases. Pediatr. Dent. 2004, 26, 277–282. [Google Scholar]
- Hanemann, J.A.C.; de Carvalho, B.C.F.; Franco, E.C. Oral manifestations in Ellis-van Creveld syndrome: Report of a case and review of the literature. J. Oral Maxillofac. Surg. 2010, 68, 456–460. [Google Scholar] [CrossRef]
- Naqash, T.A.; Alshahrani, I.; Simasetha, S. Ellis-van creveld syndrome: A rare clinical report of oral rehabilitation by interdisciplinary approach. Case Rep. Dent. 2018, 2018. [Google Scholar] [CrossRef]
- Bohnsack, B.L.; Gallina, D.; Thompson, H.; Kasprick, D.S.; Lucarelli, M.J.; Dootz, G.; Nelson, C.; McGonnell, I.M.; Kahana, A. Development of extraocular muscles requires early signals from periocular neural crest and the developing eye. Arch. Ophthalmol. 2011, 129, 1030–1041. [Google Scholar] [CrossRef]
- Versteegh, F.G.; Buma, S.A.; Costin, G.; de Jong, W.C.; Hennekam, R.C.; Ev, C.W.P. Growth hormone analysis and treatment in Ellis-van Creveld syndrome. Am. J. Med. Genet. A 2007, 143A, 2113–2121. [Google Scholar] [CrossRef]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Ornitz, D.M. Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 2013, 5, a008334. [Google Scholar] [CrossRef] [PubMed]
- Su, W.C.; Kitagawa, M.; Xue, N.; Xie, B.; Garofalo, S.; Cho, J.; Deng, C.; Horton, W.A.; Fu, X.Y. Activation of Stat1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature 1997, 386, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Sahni, M.; Ambrosetti, D.C.; Mansukhani, A.; Gertner, R.; Levy, D.; Basilico, C. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. 1999, 13, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Balmes, G.; McKinney, S.; Zhang, Z.; Givol, D.; de Crombrugghe, B. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev. 2004, 18, 290–305. [Google Scholar] [CrossRef] [PubMed]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef] [PubMed]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 1996, 273, 613–622. [Google Scholar] [CrossRef]
- Garcia, S.; Dirat, B.; Tognacci, T.; Rochet, N.; Mouska, X.; Bonnafous, S.; Patouraux, S.; Tran, A.; Gual, P.; Le Marchand-Brustel, Y.; et al. Postnatal soluble FGFR3 therapy rescues achondroplasia symptoms and restores bone growth in mice. Sci. Transl. Med. 2013, 5, 203ra124. [Google Scholar] [CrossRef]
- Pacheco, M.; Valencia, M.; Caparrós-Martín, J.A.; Mulero, F.; Goodship, J.A.; Ruiz-Perez, V.L. Evc works in chondrocytes and osteoblasts to regulate multiple aspects of growth plate development in the appendicular skeleton and cranial base. Bone 2012, 50, 28–41. [Google Scholar] [CrossRef]
- Badri, M.K.; Zhang, H.; Ohyama, Y.; Venkitapathi, S.; Alamoudi, A.; Kamiya, N.; Takeda, H.; Ray, M.; Scott, G.; Tsuji, T.; et al. Expression of Evc2 in craniofacial tissues and craniofacial bone defects in Evc2 knockout mouse. Arch. Oral Biol. 2016, 68, 142–152. [Google Scholar] [CrossRef][Green Version]
- Badri, M.K.; Zhang, H.; Ohyama, Y.; Venkitapathi, S.; Kamiya, N.; Takeda, H.; Ray, M.; Scott, G.; Tsuji, T.; Kunieda, T.; et al. Ellis Van Creveld2 is Required for Postnatal Craniofacial Bone Development. Anat. Rec. 2016, 299, 1110–1120. [Google Scholar] [CrossRef]
- Mishina, Y.; Snider, T.N. Neural crest cell signaling pathways critical to cranial bone development and pathology. Exp. Cell Res. 2015, 325, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.K.; Louie, K.a.W.; Yatabe, M.; De Oliveira Ruellas, A.C.; Mochida, Y.; Cevidanes, L.H.S.; Mishina, Y.; Zhang, H. A Ciliary Protein EVC2/LIMBIN Plays a Critical Role in the Skull Base for Mid-Facial Development. Front. Physiol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.K.; Louie, K.a.W.; Kulkarni, A.; Yatabe, M.; Carlos, A.; Ruellas, D.E.O.; Snider, T.N.; Mochida, Y.; Cevidanes, L.H.S.; Mishina, Y. The Role of Ellis-Van Creveld 2 ( EVC2 ) in Mice During Cranial Bone Development. Anat. Rec. 2018, 301, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Thesleff, I.; Hurmerinta, K. Tissue Interactions in Tooth Development. Differentiation 1981, 18, 75–88. [Google Scholar] [CrossRef]
- Zhang, H.; Takeda, H.; Tsuji, T.; Kamiya, N.; Kunieda, T.; Mochida, Y.; Mishina, Y. Loss of Function of Evc2 in Dental Mesenchyme Leads to Hypomorphic Enamel. J. Dent. Res. 2017, 96, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Ishibashi, T.; Ashizawa, H.; Shibata, T. Chondrodysplastic Dwarfism in Japanese Brown Cattle. J. Jpn. Vet. Med. Assoc. 1989, 42, 173–177. [Google Scholar] [CrossRef][Green Version]
- Yoneda, K.; Moritomo, Y.; Takami, M.; Hirata, S.; Kikukawa, Y.; Kunieda, T. Localization of a locus responsible for the bovine chondrodysplastic dwarfism (bcd) on chromosome 6. Mamm. Genome 1999, 10, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Murgiano, L.; Jagannathan, V.; Benazzi, C.; Bolcato, M.; Brunetti, B.; Muscatello, L.V.; Dittmer, K.; Piffer, C.; Gentile, A.; Drögemüller, C. Deletion in the EVC2 gene causes chondrodysplastic dwarfism in Tyrolean grey cattle. PLoS ONE 2014, 9, e0094861. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Nishimura, T.; Kuchida, K.; Mannen, H. The Japanese Wagyu beef industry: Current situation and future prospects—A review. Asian-Australas. J. Anim. Sci. 2018, 31, 933–950. [Google Scholar] [CrossRef]
- Muscatello, L.V.; Benazzi, C.; Dittmer, K.E.; Thompson, K.G.; Murgiano, L.; Drögemüller, C.; Avallone, G.; Gentile, A.; Edwards, J.F.; Piffer, C.; et al. Ellis–van Creveld Syndrome in Grey Alpine Cattle: Morphologic, Immunophenotypic, and Molecular Characterization. Vet. Pathol. 2015, 52, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, Y.; Li, J.; Kong, L.; Hu, H.; Pan, H.; Xu, L.; Deng, Y.; Li, Q.; Jin, L.; et al. Two Antarctic penguin genomes reveal insights into their evolutionary history and molecular changes related to the Antarctic environment. GigaScience 2014, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anatomical Locations | EVC | Weyers Acrodental Dysostosis | Evc Mutant Mice | Evc2/Limbin Mutant Mice | Hedgehog Signaling Defects in Mice |
|---|---|---|---|---|---|
| Limb | Short | Short (mild) | Short | Short | Short |
| Craniofacial | Normal | Normal | Normal | Normal | Cleft lips, cleft Palate |
| Neural tube | Normal | Normal | Normal | Normal | Open neural tube |
| Digit | Postaxial polydactyly | Postaxial polydactyly | Postaxial polydactyly | Postaxial polydactyly | Preaxial polydactyly (?) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Louie, K.W.; Mishina, Y.; Zhang, H. Molecular and Cellular Pathogenesis of Ellis-van Creveld Syndrome: Lessons from Targeted and Natural Mutations in Animal Models. J. Dev. Biol. 2020, 8, 25. https://doi.org/10.3390/jdb8040025
Louie KW, Mishina Y, Zhang H. Molecular and Cellular Pathogenesis of Ellis-van Creveld Syndrome: Lessons from Targeted and Natural Mutations in Animal Models. Journal of Developmental Biology. 2020; 8(4):25. https://doi.org/10.3390/jdb8040025
Chicago/Turabian StyleLouie, Ke’ale W., Yuji Mishina, and Honghao Zhang. 2020. "Molecular and Cellular Pathogenesis of Ellis-van Creveld Syndrome: Lessons from Targeted and Natural Mutations in Animal Models" Journal of Developmental Biology 8, no. 4: 25. https://doi.org/10.3390/jdb8040025
APA StyleLouie, K. W., Mishina, Y., & Zhang, H. (2020). Molecular and Cellular Pathogenesis of Ellis-van Creveld Syndrome: Lessons from Targeted and Natural Mutations in Animal Models. Journal of Developmental Biology, 8(4), 25. https://doi.org/10.3390/jdb8040025